- Metal aquo complex
-
Metal aquo complexes are coordination compounds containing metal ions with only water as a ligand. These complexes are the predominant species in aqueous solutions of many metal salts, such as metal nitrates, sulfates, and perchlorates. They have the general stoichiometry [M(H2O)n]z+. Their behavior underpins many aspects of environmental, biological, and industrial chemistry. This article focuses on complexes where water is the only ligand ("homoleptic aquo complexes"), but of course many complexes are known to consist of a mix of aquo and other ligands.[1]
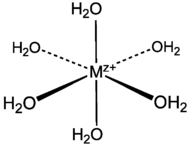 Structure of an octahedral metal aquo complex.
Structure of an octahedral metal aquo complex.
Contents
Stoichiometry and structure
Most common are the octahedral complexes with the formula [M(H2O)6]2+ and [M(H2O)6]3+. Some members of this series are listed in the table below. A few aquo complexes exist with coordination numbers lower than six. Palladium(II) and platinum(II), for example, form square planar aquo complexes with the stoichiometry [M(H2O)4]2+. Aquo complexes of the lanthanide trications are eight- and nine-coordinate, reflecting the large size of the metal centres.
Aquo complexes of about one third of the transition metals (Zr, Hf, Nb, Ta, Mo, W, Tc, Re, Os and Au) are either unknown or very rarely described. Aquo complexes of M4+ centres would be extraordinarily acidic. For example, [Ti(H2O)6]4+ is unknown, but [Ti(H2O)6]3+ is well characterized. This acidification is related to the stoichiometry of the Zr(IV) aquo complex [Zr4(OH)12(H2O)16]8+ (see zirconyl chloride. Similarly, [V(H2O)6]5+ is unknown, but its conjugate base, [VO(H2O)5]2+ is highly stable. Univalent metal centres such as Cu(I) and Rh(I) rarely form complexes with water. Ag(I) form tetrahedral [Ag(H2O)4]+, a rare example of a tetrahedral aquo complex.[2]
Some aquo complexes also contain metal-metal bond]]s. Two examples are [Mo2(H2O)8]4+ and [Rh2(H2O)10]4+.[2]
-
Complex colour electron config. M-O distance (Å)[3] water exchange
rate (s−1, 25 °C)[4]M2+/3+ self-exchange
rate (M−1s−1, 25 °C)[Ti(H2O)6]3+ violet (t2g)1 2.025 1.8 x 105 n.a. [V(H2O)6]2+ violet (t2g)3 2.12 fast [V(H2O)6]3+ green (t2g)2 1.991 fast [Cr(H2O)6]2+ blue (t2g)3(eg)1 2.06, 2.33 1.2 x 108 slow [Cr(H2O)6]3+ violet (t2g)3 1.961 2.4 x 10−6 slow [Mn(H2O)6]2+ pale pink (t2g)3(eg)2 2.177 2.1 x 107 n.a. [Fe(H2O)6]2+ blue-green (t2g)4(eg)2 2.095 4.4 x 106 fast [Fe(H2O)6]3+ pale yellow (t2g)3(eg)2 1.990 1.6 x 102 fast [Co(H2O)6]2+ pink (t2g)5(eg)2 2.08 3.2 x 106 n.a. [Ni(H2O)6]2+ green (t2g)6(eg)2 2.05 3.2 x 104 n.a. [Cu(H2O)6]2+ blue (t2g)6(eg)3 1.97, 2.30 5.7 x 109 n.a.
Reactions
Three reactions are most fundamental to the behavior of metal aquo ions: ligand exchange, electron-transfer, and acid-base reactions of the O-H bonds.
Water exchange
Ligand exchange involve replacement of a water ligand ("coordinated water") with water in solution ("bulk water"). Often the process is represented using labeled water H2O*:
- [M(H2O)n]z+ + H2O* → [M(H2O)n-1(H2O*)]z+ + H2O
In the absence of isotopic labeling, the reaction is degenerate, meaning that the free energy change is zero. Rates vary over many orders of magnitude. The main factor affecting rates is charge: highly charged metal aquo cations exchange their water more slowly than singly charged species. Thus, the exchange rates for [Na(H2O)6]+ and [Al(H2O)6]3+ differ by a factor of 109. Electron configuration is also a major factor, illustrated by the fact that the rates of water exchange for [Al(H2O)6]3+ and [Ir(H2O)6]3+ differ by a factor of 109 also.[4] Water exchange usually follows a dissociative substitution pathway, so the rate constants indicate first order reactions.
Electron exchange
This reaction usually applies to the interconversion of di- and trivalent metal ions, which involves the exchange of only one electron. The process is called self-exchange, meaning that the ion appears to exchange electrons with itself. The standard electrode potential for the equilibrium
- [M(H2O)6]2+ + [M(H2O)6]3+
 [M(H2O)6]3+ + [M(H2O)6]2+
[M(H2O)6]3+ + [M(H2O)6]2+
shows the increasing stability of the lower oxidation state as atomic number increases. The very large value for the manganese couple is a consequence of the fact that octahedral manganese(II) has zero crystal field stabilization energy (CFSE) but manganese(III) has 3 units of CFSE. [5]
Using labels to keep track of the metals, the self-exchange process is written as:
- [M(H2O)6]2+ + [M*(H2O)6]3+ → [M*(H2O)6]2+ + [M(H2O)6]3+
The rates of electron exchange vary widely, the variations being attributable to differing reorganization energies: when the 2+ and 3+ ions differ widely in structure, the rates tend to be slow.[6] The electron transfer reaction proceeds via an outer sphere electron transfer. Most often large reorganizational energies are associated with changes in the population of the eg level, at least for octahedral complexes
Acid-base reactions
Solutions of metal aquo complexes are acidic owing to the ionization of protons from the water ligands. In dilute solution chromium(III) aquo complex has a pKa of about 4.3:
- [Cr(H2O)6]3+ [Cr(H2O)5(OH)]2+ + H+
Thus, the aquo ion is a weak acid, of comparable strength to acetic acid (pKa of about 4.8). This is typical of the trivalent ions. The influence of the electronic configuration on acidity is shown by the fact that [Ru(H2O)6]3+ (pKa = 2.7) is more acidic than [Rh(H2O)6]3+ (pKa =4), despite the fact that Rh(III) is expected to be more electronegative. This effect is related to the stabilization of the pi-donor hydroxide ligand by the (t2g)5 Ru(III) centre.[2]
In more concentrated solutions, some metal hydroxo complexes undergo condensation reactions, known as olation, to form polymeric species. Many minerals, form via olation. Aquo ions of divalent metal ions are less acidic than those of trivalent cations.
The hydrolyzed species species often exhibit very different properties from the precursor hexaaquo complex. For example, water exchange in [Al(H2O)5OH]2+ is some 20000 times faster than in [Al(H2O)6]3+.
References
- ^ Mark I. Ogden and Paul D. Beer "Water & O-Donor Ligands" in Encyclopedia of Inorganic Chemistry, Wiley-VCH, 2006, Weinheim. doi:10.1002/0470862106.ia255
- ^ a b c S. F. Lincoln, D. T. Richens, A. G. Sykes "Metal Aqua Ions" Comprehensive Coordination Chemistry II Volume 1, Pages 515-555. doi:10.1016/B0-08-043748-6/01055-0
- ^ For Mn(II), Fe(II), Fe(III): T. K. Sham, J. B. Hastings, M. L. Perlman "Structure and dynamic behavior of transition-metal ions in aqueous solution: an EXAFS study of electron-exchange reactions" J. Am. Chem. Soc., 1980, 102 (18), pp 5904–5906. doi:10.1021/ja00538a033. For Ti(III), V(III), Cr(III): B. Kallies, R. Meier "Electronic Structure of 3d [M(H2O)6]3+ Ions from ScIII to FeIII: A Quantum Mechanical Study Based on DFT Computations and Natural Bond Orbital Analyses" Inorg. Chem., 2001, 40 (13), pp 3101–3112. doi:10.1021/ic001258t
- ^ a b Lothar Helm, André E. Merbach "Inorganic and Bioinorganic Solvent Exchange Mechanisms" Chemical Reviews 2005, volume 105, 1923-1959. doi:10.1021/cr030726o
- ^ Burgess, John (1978). Metal Ions in Solution. Chichester: Ellis Horwood. ISBN 0-85312-027-7. p. 236.
- ^ R. G. Wilkins Kinetics and Mechanism of Reactions of Transition Metal Complexes, 2nd Edition, VCH, Weinheim, 1991. ISBN 1-56081-125-0
Categories:- Coordination chemistry
- Inorganic chemistry
- Water chemistry
-
Wikimedia Foundation. 2010.