- Rate equation
-
The rate law or rate equation for a chemical reaction is an equation that links the reaction rate with concentrations or pressures of reactants and constant parameters (normally rate coefficients and partial reaction orders).[1] To determine the rate equation for a particular system one combines the reaction rate with a mass balance for the system.[2] For a generic reaction aA + bB → C with no intermediate steps in its reaction mechanism (that is, an elementary reaction), the rate is given by
where [A] and [B] express the concentration of the species A and B, respectively (usually in moles per liter (molarity, M)); x and y are not the respective stoichiometric coefficients of the balanced equation; they must be determined experimentally. k is the rate coefficient or rate constant of the reaction. The value of this coefficient k depends on conditions such as temperature, ionic strength, surface area of the adsorbent or light irradiation. For elementary reactions, the rate equation can be derived from first principles using collision theory. Again, x and y are NOT always derived from the balanced equation.
The rate equation of a reaction with a multi-step mechanism cannot, in general, be deduced from the stoichiometric coefficients of the overall reaction; it must be determined experimentally. The equation may involve fractional exponential coefficients, or it may depend on the concentration of an intermediate species.
The rate equation is a differential equation, and it can be integrated to obtain an integrated rate equation that links concentrations of reactants or products with time.
If the concentration of one of the reactants remains constant (because it is a catalyst or it is in great excess with respect to the other reactants), its concentration can be grouped with the rate constant, obtaining a pseudo constant: If B is the reactant whose concentration is constant, then r = k[A][B] = k'[A]. The second-order rate equation has been reduced to a pseudo-first-order rate equation. This makes the treatment to obtain an integrated rate equation much easier.
Contents
Zeroth-order reactions
A Zeroth-order reaction has a rate that is independent of the concentration of the reactant(s). Increasing the concentration of the reacting species will not speed up the rate of the reaction. Zeroth-order reactions are typically found when a material that is required for the reaction to proceed, such as a surface or a catalyst, is saturated by the reactants. The rate law for a zeroth-order reaction is
where r is the reaction rate and k is the reaction rate coefficient with units of concentration/time. If, and only if, this zeroth-order reaction 1) occurs in a closed system, 2) there is no net build-up of intermediates, and 3) there are no other reactions occurring, it can be shown by solving a mass balance equation for the system that:
If this differential equation is integrated it gives an equation often called the integrated zero-order rate law.
where
![\ [A]_t](b/a9b057049a418cfac361ebb90d56d929.png) represents the concentration of the chemical of interest at a particular time, and
represents the concentration of the chemical of interest at a particular time, and ![\ [A]_0](3/cc3b9d535081e84d36778034689277d7.png) represents the initial concentration.
represents the initial concentration.A reaction is zeroth order if concentration data are plotted versus time and the result is a straight line. The slope of this resulting line is the negative of the zero order rate constant k.
The half-life of a reaction describes the time needed for half of the reactant to be depleted (same as the half-life involved in nuclear decay, which is a first-order reaction). For a zero-order reaction the half-life is given by
- Example of a zeroth-order reaction
- Reversed Haber process:

It should be noted that the order of a reaction cannot be deduced from the chemical equation of the reaction.
First-order reactions
- See also Order of reaction.
A first-order reaction depends on the concentration of only one reactant (a unimolecular reaction). Other reactants can be present, but each will be zero-order. The rate law for an elementary reaction that is first order with respect to a reactant A is
k is the first order rate constant, which has units of 1/s.
The integrated first-order rate law is
A plot of ln [A] vs. time t gives a straight line with a slope of − k.
The half-life of a first-order reaction is independent of the starting concentration and is given by
 .
.Examples of reactions that are first-order with respect to the reactant:
Further Properties of First-Order Reaction Kinetics
The integrated first-order rate law
is usually written in the form of the exponential decay equation
A different (but equivalent) way of considering first order kinetics is as follows: The exponential decay equation can be rewritten as:
where Δtp corresponds to a specific time period and n is an integer corresponding to the number of time periods. At the end of each time period, the fraction of the reactant population remaining relative to the amount present at the start of the time period, fRP, will be:
Such that after n time periods, the fraction of the original reactant population will be:
where: fBP corresponds to the fraction of the reactant population that will break down in each time period. This equation indicates that the fraction of the total amount of reactant population that will break down in each time period is independent of the initial amount present. When the chosen time period corresponds to
 , the fraction of the population that will break down in each time period will be exactly ½ the amount present at the start of the time period (i.e. the time period corresponds to the half-life of the first-order reaction).
, the fraction of the population that will break down in each time period will be exactly ½ the amount present at the start of the time period (i.e. the time period corresponds to the half-life of the first-order reaction).The average rate of the reaction for the nth time period is given by:
Therefore, the amount remaining at the end of each time period will be related to the average rate of that time period and the reactant population at the start of the time period by:
- An = An − 1 − ravg,nΔtp
Since the fraction of the reactant population that will break down in each time period can be expressed as:
The amount of reactant that will break down in each time period can be related to the average rate over that time period by:
Such that the amount that remains at the end of each time period will be related to the amount present at the start of the time period according to:
This equation is a recursion allowing for the calculation of the amount present after any number of time periods, without need of the rate constant, provided that the average rate for each time period is known. [3]
Second-order reactions
A second-order reaction depends on the concentrations of one second-order reactant, or two first-order reactants.
For a second order reaction, its reaction rate is given by:
![\ -\frac{d[A]}{dt} = 2k[A]^2](3/873a3b7d216517de98cb8ee3a86ceb44.png) or
or ![\ -\frac{d[A]}{dt} = k[A][B]](d/cfd5938b3763344a202d6975a0d68ed9.png) or
or ![\ -\frac{d[A]}{dt} = 2k[B]^2](1/781cda9b25d2e92fb50c2f6a30188f41.png)
In several popular kinetics books, the definition of the rate law for second-order reactions is
![-\frac{d[A]}{dt} = k[A]^2](c/58cd77b68b6d8db6df381fc176b25a1d.png) . Conflating the 2 inside the constant for the first, derivative, form will only make it required in the second, integrated form (presented below). The option of keeping the 2 out of the constant in the derivative form is considered more correct, as it is almost always used in peer-reviewed literature, tables of rate constants, and simulation software.[4]
. Conflating the 2 inside the constant for the first, derivative, form will only make it required in the second, integrated form (presented below). The option of keeping the 2 out of the constant in the derivative form is considered more correct, as it is almost always used in peer-reviewed literature, tables of rate constants, and simulation software.[4]The integrated second-order rate laws are respectively
or
[A]0 and [B]0 must be different to obtain that integrated equation.
The half-life equation for a second-order reaction dependent on one second-order reactant is
![\ t_ \frac{1}{2} = \frac{1}{k[A]_0}](e/9ce65475b614802ec855072f11df0d99.png) . For a second-order reaction half-lives progressively double.
. For a second-order reaction half-lives progressively double.Another way to present the above rate laws is to take the log of both sides:
![\ln{}r = \ln{}k + 2\ln\left[A\right]](0/cf04309d2e7d76fc386ebfcf6877723f.png)
- Examples of a Second-order reaction
Pseudo-first-order
Measuring a second-order reaction rate with reactants A and B can be problematic: The concentrations of the two reactants must be followed simultaneously, which is more difficult; or measure one of them and calculate the other as a difference, which is less precise. A common solution for that problem is the pseudo-first-order approximation
If either [A] or [B] remains constant as the reaction proceeds, then the reaction can be considered pseudo-first-order because, in fact, it depends on the concentration of only one reactant. If, for example, [B] remains constant, then:
![\ r = k[A][B] = k'[A]](4/7b4a55d51c83a671365d244987234a56.png)
where k' = k[B]0 (k' or kobs with units s−1) and an expression is obtained identical to the first order expression above.
One way to obtain a pseudo-first-order reaction is to use a large excess of one of the reactants ([B]>>[A] would work for the previous example) so that, as the reaction progresses, only a small amount of the reactant is consumed, and its concentration can be considered to stay constant. By collecting k' for many reactions with different (but excess) concentrations of [B], a plot of k' versus [B] gives k (the regular second order rate constant) as the slope.
Example: The hydrolysis of esters by dilute mineral acids follows pseudo-first-order kinetics where the concentration of water is present in large excess.
- CH3COOCH3 + H2O → CH3COOH + CH3OH
Summary for reaction orders 0, 1, 2, and n
Elementary reaction steps with order 3 (called ternary reactions) are rare and unlikely to occur. However, overall reactions composed of several elementary steps can, of course, be of any (including non-integer) order.
Zero-Order First-Order Second-Order nth-Order Rate Law ![-\frac{d[A]}{dt} = k](0/b3002675ed0c609a10157d0c733543b0.png)
![-\frac{d[A]}{dt} = k[A]](0/e802dbfb8a252f2bf0c9729a4e6e66bb.png) [4]
[4]![-\frac{d[A]}{dt} = k[A]^n](f/36f83d341091d001bf230ba93dc66181.png)
Integrated Rate Law ![\ [A] = [A]_0 - kt](f/86fcee80fab8997ba85af1ff815abeff.png)
![\ [A] = [A]_0 e^{-kt}](a/f3acb1952af0a546a0c3162b0c262341.png)
![\frac{1}{[A]} = \frac{1}{[A]_0} + kt](4/664842f28db997704b7ddbc61f6a7be8.png) [4]
[4]![\frac{1}{[A]^{n-1}} = \frac{1}{{[A]_0}^{n-1}} + (n-1)kt](4/ed46a209d9c598bad5186d11c6834122.png)
[Except first order]
Units of Rate Constant (k) 



Linear Plot to determine k ![[A] \ \mbox{vs.} \ t](a/b8a126629e29133c39b43abc6d175bcd.png)
![\ln ([A]) \ \mbox{vs.} \ t](8/bc887676255e78b0311b24a38cdca806.png)
![\frac{1}{[A]} \ \mbox{vs.} \ t](7/4d793f30055b332220dc5c0cc98e394c.png)
![\frac{1}{[A]^{n-1}} \ \mbox{vs.} \ t](b/3ebf4ce732399ed4911ed113b04e5b8a.png)
[Except first order]
Half-life ![t_{1/2} = \frac{[A]_0}{2k}](e/efe758e6239079a41ba9d39d5cc395c2.png)

![t_{1/2} = \frac{1}{k[A]_0}](f/dbff097ba7cc49335837f67b29a25f9a.png) [4]
[4]![t_{1/2} = \frac{2^{n-1}-1}{(n-1)k{[A]_0}^{n-1}}](4/164c7ff91d8653c5cccdb2ff22e78a61.png)
[Except first order]
Where M stands for concentration in molarity (mol · L−1), t for time, and k for the reaction rate constant. The half-life of a first-order reaction is often expressed as t1/2 = 0.693/k (as ln2 = 0.693).
Equilibrium reactions or opposed reactions
A pair of forward and reverse reactions may define an equilibrium process. For example, A and B react into X and Y and vice versa (s, t, u, and v are the stoichiometric coefficients):
The reaction rate expression for the above reactions (assuming each one is elementary) can be expressed as:
where: k1 is the rate coefficient for the reaction that consumes A and B; k2 is the rate coefficient for the backwards reaction, which consumes X and Y and produces A and B.
The constants k1 and k2 are related to the equilibrium coefficient for the reaction (K) by the following relationship (set r=0 in balance):
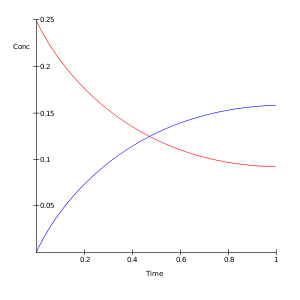 Concentration of A (A0 = 0.25 mole/l) and B versus time reaching equilibrium kf = 2 min-1 and kr = 1 min-1
Concentration of A (A0 = 0.25 mole/l) and B versus time reaching equilibrium kf = 2 min-1 and kr = 1 min-1
Simple Example
In a simple equilibrium between two species:
Where the reactions starts with an initial concentration of A, [A]0, with an initial concentration of 0 for B at time t=0.
Then the constant K at equilibrium is expressed as:
Where [A]e and [B]e are the concentrations of A and B at equilibrium, respectively.
The concentration of A at time t, [A]t, is related to the concentration of B at time t, [B]t, by the equilibrium reaction equation:
Note that the term [B]0 is not present because, in this simple example, the initial concentration of B is 0.
This applies even when time t is at infinity; i.e., equilibrium has been reached:
then it follows, by the definition of K, that
and, therefore,
These equations allow us to uncouple the system of differential equations, and allow us to solve for the concentration of A alone.
The reaction equation, given previously as:
The derivative is negative because this is the rate of the reaction going from A to B, and therefore the concentration of A is decreasing. To simplify annotation, let x be [A]t, the concentration of A at time t. Let xe be the concentration of A at equilibrium. Then:
Since:
The reaction rate becomes:
which results in:
A plot of the negative natural logarithm of the concentration of A in time minus the concentration at equilibrium versus time t gives a straight line with slope kf + kb. By measurement of Ae and Be the values of K and the two reaction rate constants will be known.[5]
Generalization of Simple Example
If the concentration at the time t = 0 is different from above, the simplifications above are invalid, and a system of differential equations must be solved. However, this system can also be solved exactly to yield the following generalized expressions:
![\left[ A \right]=\left[ A \right]_{0}\frac{1}{k_{f}+k_{b}}\left( k_{b}+k_{f}e^{-\left( k_{f}+k_{b} \right)t} \right)+\left[ B \right]_{0}\frac{k_{b}}{k_{f}+k_{b}}\left( 1-e^{-\left( k_{f}+k_{b} \right)t} \right)](e/41ea08d07c8ef40bb1a45bafe35b8499.png)
![\left[ B \right]=\left[ A \right]_{0}\frac{k_{f}}{k_{f}+k_{b}}\left( 1-e^{-\left( k_{f}+k_{b} \right)t} \right)+\left[ B \right]_{0}\frac{1}{k_{f}+k_{b}}\left( k_{f}+k_{b}e^{-\left( k_{f}+k_{b} \right)t} \right)](e/5cef68de8e3613120123dfe76fd51380.png)
When the equilibrium constant is close to unity and the reaction rates very fast for instance in conformational analysis of molecules, other methods are required for the determination of rate constants for instance by complete lineshape analysis in NMR spectroscopy.
Consecutive reactions
If the rate constants for the following reaction are k1 and k2;
 , then the rate equation is:
, then the rate equation is:For reactant A:
![\frac{d[A]}{dt} = -k_1 [A]](e/86e24b62bbf0b6d0c81b6ff5143b8ab9.png)
For reactant B:
![\frac{d[B]}{dt} = k_1 [A] - k_2 [B]](b/ddbf39f88d2aea78c65e997ae87434ae.png)
For product C:
![\frac{d[C]}{dt} = k_2 [B]](b/39b9e2213ec97994c460c034d38c59ae.png)
With the individual concentrations scaled by the total population of reactants to become probabilities, linear systems of differential equations such as these can be formulated as a master equation. The differential equations can be solved analytically and the integrated rate equations are
![[A]=[A]_0 e^{-k_1 t}](5/7256d2c1532cc5805adcedb96279b69a.png)
![\left[ B \right]=\left\{ \begin{array}{*{35}l}
\left[ A \right]_{0}\frac{k_{1}}{k_{2}-k_{1}}\left( e^{-k_{1}t}-e^{-k_{2}t} \right)+\left[ B \right]_{0}e^{-k_{2}t} & k_{1}\ne k_{2} \\
\left[ A \right]_{0}k_{1}te^{-k_{1}t}+\left[ B \right]_{0}e^{-k_{1}t} & \text{otherwise} \\
\end{array} \right.](3/623a5d8b2b965a49f6540f7c9ba49c7e.png)
![\left[ C \right]=\left\{ \begin{array}{*{35}l}
\left[ A \right]_{0}\left( 1+\frac{k_{1}e^{-k_{2}t}-k_{2}e^{-k_{1}t}}{k_{2}-k_{1}} \right)+\left[ B \right]_{0}\left( 1-e^{-k_{2}t} \right)+\left[ C \right]_{0} & k_{1}\ne k_{2} \\
\left[ A \right]_{0}\left( 1-e^{-k_{1}t}-k_{1}te^{-k_{1}t} \right)+\left[ B \right]_{0}\left( 1-e^{-k_{1}t} \right)+\left[ C \right]_{0} & \text{otherwise} \\
\end{array} \right.](f/d1fce437ed200540890e888f46df1e0a.png)
The steady state approximation leads to very similar results in an easier way.
Parallel or competitive reactions
When a substance reacts simultaneously to give two different products, a parallel or competitive reaction is said to take place.
- Two first order reactions:
 and
and  , with constants k1 and k2 and rate equations
, with constants k1 and k2 and rate equations ![-\frac{d[A]}{dt}=(k_1+k_2)[A]](4/d04394a9bfcd42230c3e1381fec71d82.png) ,
, ![\frac{d[B]}{dt}=k_1[A]](c/70c1dea62ee39ded884e987e5c9aedf3.png) and
and ![\frac{d[C]}{dt}=k_2[A]](b/a0b010b90ca7de616420b99117f81369.png)
The integrated rate equations are then
![\ [A] = [A]_0 e^{-(k_1+k_2)t}](f/5fff2656e4c0d91000034a78835cd799.png) ;
; ![[B] = \frac{k_1}{k_1+k_2}[A]_0 (1-e^{-(k_1+k_2)t})](f/98f477373e35c5723c85bc5629c32569.png) and
and ![[C] = \frac{k_2}{k_1+k_2}[A]_0 (1-e^{-(k_1+k_2)t})](5/0953b7867deff6def848ed0b236d7709.png) .
.One important relationship in this case is
![\frac{[B]}{[C]}=\frac{k_1}{k_2}](4/ad4a86d319ac3e8f781d1c8e075422ae.png)
- One first order and one second order reaction:[6]
This can be the case when studying a bimolecular reaction and a simultaneous hydrolysis (which can be treated as pseudo order one) takes place: the hydrolysis complicates the study of the reaction kinetics, because some reactant is being "spent" in a parallel reaction. For example A reacts with R to give our product C, but meanwhile the hydrolysis reaction takes away an amount of A to give B, a byproduct:
 and
and  . The rate equations are:
. The rate equations are: ![\frac{d[B]}{dt}=k_1[A][H_2O]=k_1'[A]](e/cbef138ce847e5fa0a11569b9739d693.png) and
and ![\frac{d[C]}{dt}=k_2[A][R]](4/c146186b77712d9ace92327a4e91dd4b.png) . Where k1' is the pseudo first order constant.
. Where k1' is the pseudo first order constant.The integrated rate equation for the main product [C] is
![[C]=[R]_0 \left [ 1-e^{-\frac{k_2}{k_1'}[A]_0(1-e^{-k_1't})} \right ]](4/2e4456d93007140e9859124322dabbc6.png) , which is equivalent to
, which is equivalent to ![ln \frac{[R]_0}{[R]_0-[C]}=\frac{k_2[A]_0}{k_1'}(1-e^{-k_1't})](6/df6c7da387064a2808460356cedee5ac.png) . Concentration of B is related to that of C through
. Concentration of B is related to that of C through ![[B]=-\frac{k_1'}{k_2} ln \left ( 1 - \frac{[C]}{[R]_0} \right )](a/e1a1502f77a0da87232864d40a484f38.png)
The integrated equations were analytically obtained but during the process it was assumed that
![[A]_0-[C]\approx \;[A]_0](b/84b23426a8836de817b0afce4d68e703.png) therefeore, previous equation for [C] can only be used for low concentrations of [C] compared to [A]0
therefeore, previous equation for [C] can only be used for low concentrations of [C] compared to [A]0See also
- Reaction rate
- Reaction rate constant
- Reactions on surfaces: rate equations for reactions where at least one of the reactants adsorbs onto a surface
- Reaction-diffusion equation
- Steady state approximation
References
- ^ IUPAC Gold Book definition of rate law. See also: According to IUPAC Compendium of Chemical Terminology.
- ^ Kenneth A. Connors Chemical Kinetics, the study of reaction rates in solution, 1991, VCH Publishers. This book contains most of the rate equations in this article and their derivation.
- ^ Walsh R, Martin E, Darvesh S. A method to describe enzyme-catalyzed reactions by combining steady state and time course enzyme kinetic parameters... Biochim Biophys Acta. 2010 Jan;1800:1-5
- ^ a b c d NDRL Radiation Chemistry Data Center. See also: Christos Capellos and Bennon H. Bielski "Kinetic systems: mathematical description of chemical kinetics in solution" 1972, Wiley-Interscience (New York).
- ^ For a worked out example see: Determination of the Rotational Barrier for Kinetically Stable Conformational Isomers via NMR and 2D TLC An Introductory Organic Chemistry Experiment Gregory T. Rushton, William G. Burns, Judi M. Lavin, Yong S. Chong, Perry Pellechia, and Ken D. Shimizu J. Chem. Educ. 2007, 84, 1499. Abstract
- ^ José A. Manso et al."A Kinetic Approach to the Alkylating Potential of Carcinogenic Lactones" Chem. Res. Toxicol. 2005, 18, (7) 1161-1166
Basic reaction mechanisms Nucleophilic substitution Elimination reaction Unimolecular elimination (E1) · E1cB elimination reaction · Bimolecular elimination (E2)Related topics Chemical kinetics Rate equation · Rate-determining stepCategories:- Chemical reactions
- Chemical kinetics
- Chemical engineering
![r\; =\; k[\mathrm{A}]^x[\mathrm{B}]^y](a/23a06c4ee205d7ea62ff463805c6b085.png)

![r = -\frac{d[A]}{dt}=k](6/426104668a2d1fe61fe4dbbb190d75f7.png)
![\ [A]_t = -kt + [A]_0](c/ccc8f109cf83a4114b3c6e547e95e823.png)
![\ t_ \frac{1}{2} = \frac{[A]_0}{2k}](4/6e4b883bbbf7a967b488081d26589d73.png)
![r = -\frac{d[A]}{dt} = k[A]](a/18ab6ebf0129343111def7906bba042d.png)
![\ \ln{[A]} = -kt + \ln{[A]_0}](c/76cf4348c28e3a4f00bb9c20da4e1387.png)











![\frac{[A]}{[B]} = \frac{[A]_0}{[B]_0} e^{([A]_0 - [B]_0)kt}](6/0261d2c287f23e6971bba8e5cdf68b22.png)


![r = {k_1 [A]^s[B]^t} - {k_2 [X]^u[Y]^v}\,](f/57fac86d71637ea80bcdf8bad7a84b43.png)
![{k_1 [A]^s[B]^t = k_2 [X]^u[Y]^v}\,](4/0f490754c2af17cdc6442dd2792ec008.png)
![K = \frac{[X]^u[Y]^v}{[A]^s[B]^t} = \frac{k_1}{k_2}](9/a097aa22a4f852d3180cca18a45e2a04.png)


![K \ \stackrel{\mathrm{def}}{=}\ \frac{k_{f}}{k_{b}} = \frac{\left[B\right]_e} {\left[A\right]_e}](b/05b33bde8d504ac56641d440c1e39e92.png)
![\ [A]_t = [A]_0 - [B]_t](1/7217b6ec6d58489f5e0d9a76a8104d29.png)
![\ [A]_e = [A]_0 - [B]_e](e/80e05541a7318abe5e02f4e734015214.png)
![\ [B]_e = x = \frac{k_{f}}{k_f+k_b}[A]_0](3/dd384e4a0d0a098e347b9c003c53c72c.png)
![\ [A]_e = [A]_0 - x = \frac{k_{b}}{k_f+k_b}[A]_0](4/67426c3e34401c036f4eed1af9bf1ae9.png)
![-\frac{d[A]}{dt} = {k_f [A]_t} - {k_b [B]_t}\,](d/23d6558aa6e3a0fc83cb1cfea7291c99.png)
![-\frac{dx}{dt} = {k_f x} - {k_b [B]_t}\,](9/579ab722b19eb9e396587f3e8d13d53f.png)
![-\frac{dx}{dt} = {k_f x} - {k_b ([A]_0 - x)}\,](6/726cb10764404ea8776238b01830842e.png)
![-\frac{dx}{dt} = {(k_f + k_b)x} - {k_b [A]_0}\,](1/2d179eb41932138a089062ca2f55eb49.png)
![k_f + k_b = {k_b \frac{[A]_0}{x_e}}](e/76e84bf30a4a28137f6d82941924ccc2.png)
![\ \frac{dx}{dt} = \frac{k_b[A]_0}{x_e} (x_e - x)](4/3748af0e1f40d511ef13a83b858e9116.png)
![\ln(\frac{[A]_0 - [A]_e}{[A]_t-[A]_e}) = (k_f + k_b)t](9/29979d0e9b2e92012ab2dc54174510f7.png)
Wikimedia Foundation. 2010.