- More O'Ferrall-Jencks plot
-
More O’Ferrall-Jencks plots are two-dimensional representations of multiple reaction coordinate potential energy surfaces for chemical reactions that involve simultaneous changes in two bonds. As such, they are a useful tool to explain or predict how changes in the reactants or reaction conditions can affect the position and geometry of the transition state of a reaction for which there are possible competing pathways.[1]
Contents
Brief history
These plots were first introduced by R. A. More O’Ferrall to discuss mechanisms of β-eliminations[2] and later adopted by W. P. Jencks in an attempt to clarify the finer details involved in the general acid-base catalysis of reversible addition reactions to carbon electrophiles such as the hydration of carbonyls.[3]
Description
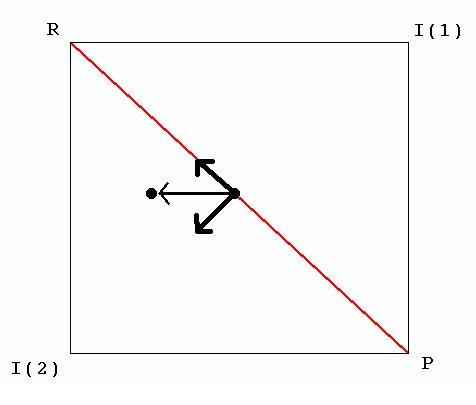
 Figure 1. A generic More O’Ferrall-Jencks plot. R, I(1), I(2) and P stand for reactant(s), intermediate(S) 1, intermediate(s) 2 and product(s) respectively. The thick arrows represent movement of the transtitions state (black dot) parallel and perpendicular to the diagonal (red line). The thin arrow is the vector sum of the thick arrows.
Figure 1. A generic More O’Ferrall-Jencks plot. R, I(1), I(2) and P stand for reactant(s), intermediate(S) 1, intermediate(s) 2 and product(s) respectively. The thick arrows represent movement of the transtitions state (black dot) parallel and perpendicular to the diagonal (red line). The thin arrow is the vector sum of the thick arrows.
In this type of plot (Figure 1), each axis represents a unique reaction coordinate, the corners represent local minima along the potential surface such as reactants, products or intermediates and the energy axis projects vertically out of the page. Changing a single reaction parameter can change the height of one or more of the corners of the plot. These changes are transmitted across the surface such that the position of the transition state (the saddle point) is altered.[4]
Consider a generic example in which the initial transition state along a concerted pathway is represented by a black dot on a red diagonal (Figure 1). Changing the height of the corners can have two effects on the position of the transition state: it can move along the diagonal, reflecting a change in the Gibbs free energy of the reaction (ΔG°), or perpendicular to it, reflecting a change in the energy of competing pathways. Thus, in accordance with the Hammond postulate, the transition state moves along the diagonal towards the corner that is raised in energy (a Hammond effect) and perpendicular to the diagonal towards the corner that is lowered (an anti-Hammond effect).[5][6] In this example, R is raised in energy and I(1) is lowered in energy. The transition state moves accordingly and the vector sum of both movements gives the real change in its position.
Applications
Elimination reactions
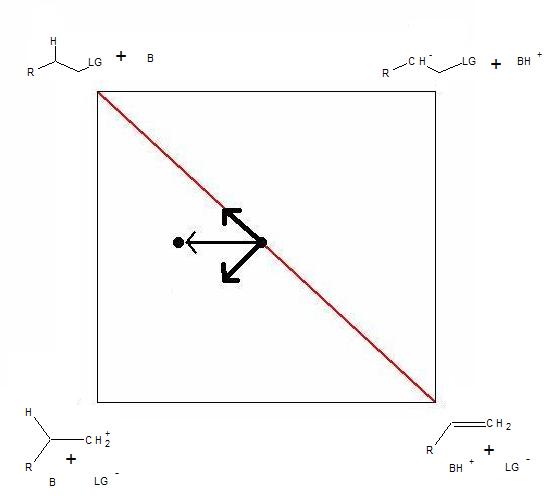
 Figure 2. More O’Ferrall-Jencks plot of the competing β-elimination mechanisms: E2, E1 and E1cB. The arrows indicate the effects of increasing the leaving group ability on the position of the transition state.
Figure 2. More O’Ferrall-Jencks plot of the competing β-elimination mechanisms: E2, E1 and E1cB. The arrows indicate the effects of increasing the leaving group ability on the position of the transition state.Initially, More O’Ferrall introduced this type of analysis to discuss the continuity between concerted and step-wise β-elimination reaction mechanisms. The model also provided a framework within which to explain the effects of substituents and reaction conditions on the mechanism.[7] The appropriate lower energy species were placed at the corners of the two dimensional plot (Figure 2). These were the reactants (top left), the products (bottom right) and the intermediates of the two possible stepwise reactions: the carbocation for E1 (bottom left) and the carbanion for E1cB (top-right). Thus, the horizontal axes represent the extent of deprotonation (C-H bond distance) and the vertical axes represent the extent of leaving group departure (C-LG distance). By applying the Hammond and anti-Hammond effects,[8] he predicted the effects of various changes in the reactants or reaction conditions. For example, the effects of introducing a better leaving group on a substrate that initially eliminates via an E2 mechanism are illustrated in Figure 2. A better leaving group increases the energy of the reactants and of the carbanion intermediate. Thus, the transition state moves towards the reactants and away from the carbanion intermediate.
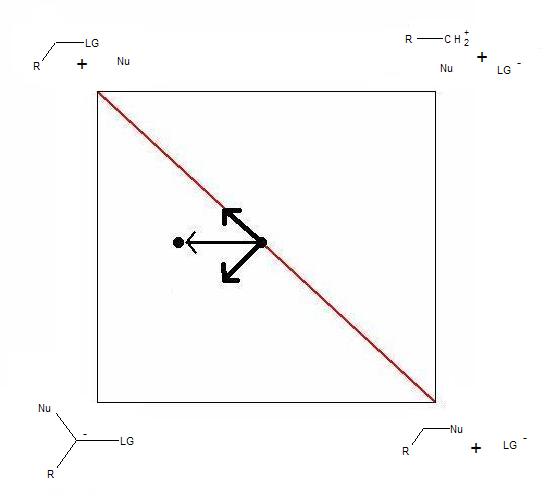
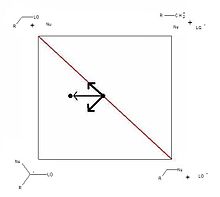 Figure 3. More O’Ferrall-Jencks plot of competing nucleophilic aliphatic substitution mechanisms: SN1 and SN2. The arrows represent the effect of increasing the nucleophilicity of the nucleophile on the position of the transition state.
Figure 3. More O’Ferrall-Jencks plot of competing nucleophilic aliphatic substitution mechanisms: SN1 and SN2. The arrows represent the effect of increasing the nucleophilicity of the nucleophile on the position of the transition state.Interestingly, the model does not predict any change in leaving group departure at the transition state. Instead the extent of deprotonation is expected to decrease. This can be explained by the fact that a better leaving group needs less assistance from a developing neighbouring negative charge in order to depart. The true change predicts more carbocation character at the transition state and a mechanism that is more E1-like. These observations can be correlated with Hammett ρ-values.[9] Poor leaving groups correlate with large positive ρ-values. Gradually increasing the leaving group ability decreases the ρ-value until it becomes large and negative, indicating the development of positive charge in the transition state.
Substitution reactions
A similar analysis, done by J. M. Harris, has been applied to the competing SN1 and SN2 nucleophilic aliphatic substitution pathways.[10] The effects of increasing the nucleophilicity of the nucleophile are shown as an example in Figure 3. An agreement with Hammet ρ-values is also apparent in this application.[11]
Addition to carbonyls
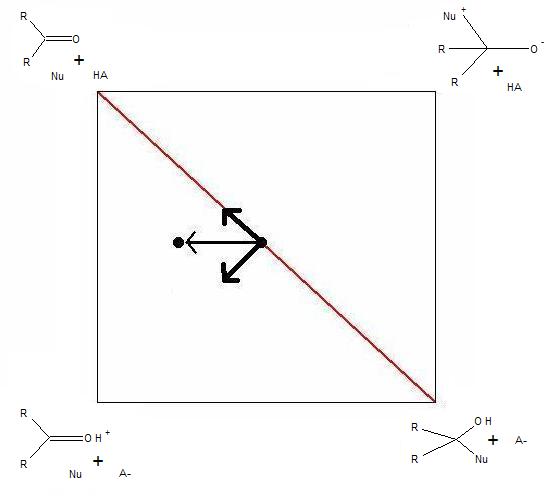
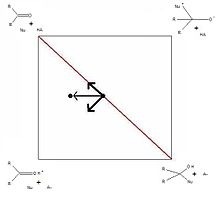 Figure 4. More O’Ferrall-Jencks plot of acid-catalyzed versus uncatalyzed nucleophilic addition to a carbonyl. The arrows represent the effects of increasing the strength of the acid on the position of the transition state.
Figure 4. More O’Ferrall-Jencks plot of acid-catalyzed versus uncatalyzed nucleophilic addition to a carbonyl. The arrows represent the effects of increasing the strength of the acid on the position of the transition state.Finally, this type of plot can readily be drawn to illustrate the effects of changing parameters in the acid-catalyzed nucleophilic addition to carbonyls. The example in Figure 4 demonstrates the effects of increasing the strength of the acid. In this case, the extent of protonation is the α-value in the Brønsted catalysis equation. The fact that the α-value remains unchanged explains the linearity of Brønsted plots for such a reaction.[12]
Ultimately, the More O’Ferrall-Jencks plots have qualitative predictive and explanatory power regarding the effects of changing substituents and reaction conditions for a wide variety of reactions.
References
- ^ Anslyn, E. V., and Dougherty, D. A. Modern Physical Organic Chemistry. California: University Science Books, 2006, pp. 407–410.
- ^ More O’Ferrall, R. A. (1970) J. Chem. Soc. B, 274.
- ^ Jencks, W. P. (1972) Chem Rev. 72, 705.
- ^ Anslyn, E. V., and Dougherty, D. A. Modern Physical Organic Chemistry. California: University Science Books, 2006, pp. 407–410.
- ^ Anslyn, E. V., and Dougherty, D. A. Modern Physical Organic Chemistry. California: University Science Books, 2006, pp. 407–410.
- ^ Thornton, E. R. (1967) J. Am. Chem. Soc. 89, 2915
- ^ More O’Ferrall, R. A. (1970) J. Chem. Soc. B, 274.
- ^ Thornton, E. R. (1967) J. Am. Chem. Soc. 89, 2915
- ^ Anslyn, E. V., and Dougherty, D. A. Modern Physical Organic Chemistry. California: University Science Books, 2006, pp. 586–588.
- ^ Harris, J. M., Shafer, S. G., Moffatt, J. E., and Becker, A. R. (1979) J. Am. Chem. Soc. 101, 3295.
- ^ Anslyn, E. V., and Dougherty, D. A. Modern Physical Organic Chemistry. California: University Science Books, 2006, p. 650.
- ^ Anslyn, E. V., and Dougherty, D. A. Modern Physical Organic Chemistry. California: University Science Books, 2006, pp. 543–544.
See also
Categories:- Plots (graphics)
- Chemical kinetics
- Physical organic chemistry
Wikimedia Foundation. 2010.