- SNP genotyping
-
SNP genotyping is the measurement of genetic variations of single nucleotide polymorphisms (SNPs) between members of a species. It is a form of genotyping, which is the measurement of more general genetic variation. SNPs are one of the most common types of genetic variation. An SNP is a single base pair mutation at a specific locus, usually consisting of two alleles (where the rare allele frequency is >1%). SNPs are found to be involved in the etiology of many human diseases and are becoming of particular interest in pharmacogenetics. Because SNPs are conserved during evolution, they have been proposed as markers for use in quantitative trait loci (QTL) analysis and in association studies in place of microsatellites. The use of SNPs is being extended in the HapMap project, which aims to provide the minimal set of SNPs needed to genotype the human genome. SNPs can also provide a genetic fingerprint for use in identity testing (Rapley & Harbron 2004). The increase in interest in SNPs has been reflected by the furious development of a diverse range of SNP genotyping methods.
Contents
Hybridization-based methods
Several applications have been developed that interrogate SNPs by hybridizing complementary DNA probes to the SNP site. The challenge of this approach is reducing cross-hybridization between the allele-specific probes. This challenge is generally overcome by manipulating the hybridization stringency conditions (Rapley & Harbron 2004).
Dynamic allele-specific hybridization
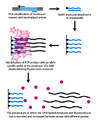

Dynamic allele-specific hybridization (DASH) genotyping takes advantage of the differences in the melting temperature in DNA that results from the instability of mismatched base pairs. The process can be vastly automated and encompasses a few simple principles.
In the first step, a genomic segment is amplified and attached to a bead through a PCR reaction with a biotinylated primer. In the second step, the amplified product is attached to a streptavidin column and washed with NaOH to remove the unbiotinylated strand. An allele-specific oligonucleotide is then added in the presence of a molecule that fluoresces when bound to double-stranded DNA. The intensity is then measured as temperature is increased until the Tm can be determined. A SNP will result in a lower than expected Tm (Howell et al. 1999).
Because DASH genotyping is measuring a quantifiable change in Tm, it is capable of measuring all types of mutations, not just SNPs. Other benefits of DASH include its ability to work with label free probes and its simple design and performance conditions.
Molecular beacons

SNP detection through molecular beacons makes use of a specifically engineered single-stranded oligonucleotide probe. The oligonucleotide is designed such that there are complementary regions at each end and a probe sequence located in between. This design allows the probe to take on a hairpin, or stem-loop, structure in its natural, isolated state. Attached to one end of the probe is a fluorophore and to the other end a fluorescence quencher. Because of the stem-loop structure of the probe, the fluorophore is in close proximity to the quencher, thus preventing the molecule from emitting any fluorescence. The molecule is also engineered such that only the probe sequence is complementary to the genomic DNA that will be used in the assay (Abravaya et al. 2003).
If the probe sequence of the molecular beacon encounters its target genomic DNA during the assay, it will anneal and hybridize. Because of the length of the probe sequence, the hairpin segment of the probe will be denatured in favour of forming a longer, more stable probe-target hybrid. This conformational change permits the fluorophore and quencher to be free of their tight proximity due to the hairpin association, allowing the molecule to fluoresce.
If on the other hand, the probe sequence encounters a target sequence with as little as one non-complementary nucleotide, the molecular beacon will preferentially stay in its natural hairpin state and no fluorescence will be observed, as the fluorophore remains quenched.
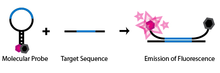
The unique design of these molecular beacons allows for a simple diagnostic assay to identify SNPs at a given location. If a molecular beacon is designed to match a wild-type allele and another to match a mutant of the allele, the two can be used to identify the genotype of an individual. If only the first probe’s fluorophore wavelength is detected during the assay then the individual is homozygous to the wild type. If only the second probe’s wavelength is detected then the individual is homozygous to the mutant allele. Finally, if both wavelengths are detected, then both molecular beacons must be hybridizing to their complements and thus the individual must contain both alleles and be heterozygous.
SNP microarrays
In high-density oligonucleotide SNP arrays, hundreds of thousands of probes are arrayed on a small chip, allowing for many SNPs to be interrogated simultaneously (Rapley & Harbron 2004). Because SNP alleles only differ in one nucleotide and because it is difficult to achieve optimal hybridization conditions for all probes on the array, the target DNA has the potential to hybridize to mismatched probes. This is addressed somewhat by using several redundant probes to interrogate each SNP. Probes are designed to have the SNP site in several different locations as well as containing mismatches to the SNP allele. By comparing the differential amount of hybridization of the target DNA to each of these redundant probes, it is possible to determine specific homozygous and heterozygous alleles (Rapley & Harbron 2004). Although oligonucleotide microarrays have a comparatively lower specificity and sensitivity, the scale of SNPs that can be interrogated is a major benefit. The Affymetrix Human SNP 5.0 GeneChip performs a genome-wide assay that can genotype over 500,000 human SNPs (Affymetrix 2007).
Enzyme-based methods
A broad range of enzymes including DNA ligase, DNA polymerase and nucleases have been employed to generate high-fidelity SNP genotyping methods.
Restriction fragment length polymorphism
Restriction fragment length polymorphism (RFLP) is considered to be the simplest and earliest method to detect SNPs. SNP-RFLP makes use of the many different restriction endonucleases and their high affinity to unique and specific restriction sites. By performing a digestion on a genomic sample and determining fragment lengths through a gel assay it is possible to ascertain whether or not the enzymes cut the expected restriction sites. A failure to cut the genomic sample results in an identifiably larger than expected fragment implying that there is a mutation at the point of the restriction site which is rendering it protected from nuclease activity.
Unfortunately, the combined factors of the high complexity of most eukaryotic genomes, the requirement for specific endonucleases, the fact that the exact mutation cannot be necessarily be resolved in a single experiment, and the slow nature of gel assays make RFLP a poor choice for high throughput analysis.
PCR-based methods
Tetra-primer ARMS-PCR employs two pairs of primers to amplify two alleles in one PCR reaction. The primers are designed such that the two primer pairs overlap at a SNP location but each match perfectly to only one of the possible SNPs. As a result, if a given allele is present in the PCR reaction, the primer pair specific to that allele will produce product but not to the alternative allele with a different SNP. The two primer pairs are also designed such that their PCR products are of a significantly different length allowing for easily distinguishable bands by gel electrophoresis.
In examining the results, if a genomic sample is homozygous, then the PCR products that result will be from the primer which matches the SNP location to the outer, opposite strand primer as well from the two opposite, outer primers. If the genomic sample is heterozygous, then products will result from the primer of each allele to their respective outer primer counterparts as well as from the two opposite, outer primers.
The difficulty in designing multiple pairs of primers for a single PCR reaction is vastly outweighed by the simplicity and speed at which samples can be examined.
Flap endonuclease
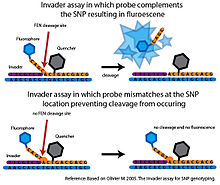
Flap endonuclease (FEN) is an endonuclease that catalyzes structure-specific cleavage. This cleavage is highly sensitive to mismatches and can be used to interrogate SNPs with a high degree of specificity (Olivier 2005).
In the basic Serial Invasive Signal Amplification Reaction (SISAR) allows both SNP alleles to be interrogated in a single reaction. SISAR Invader assay also requires less target DNA, improving the sensitivity of the original Invader assay (Olivier 2005). The assay has also been adapted in several ways for use in a high-throughput format. In one platform, the allele-specific probes are anchored to microspheres. When cleavage by FEN generates a detectable fluorescent signal, the signal is measured using flow-cytometry. The sensitivity of flow-cytometry, eliminates the need for PCR amplification of the target DNA (Rao et al. 2003). These high-throughput platforms have not progressed beyond the proof-of-principle stage and so far the Invader system has not been used in any large scale SNP genotyping projects (Olivier 2005).
Primer extension
Primer extension is a two step process that first involves the hybridization of a probe to the bases immediately upstream of the SNP nucleotide followed by a ‘mini-sequencing’ reaction, in which DNA polymerase extends the hybridized primer by adding a base that is complementary to the SNP nucleotide. This incorporated base is detected and determines the SNP allele (Goelet et al. 1999; Syvanen 2001). Because primer extension is based on the highly accurate DNA polymerase enzyme, the method is generally very reliable. Primer extension is able to genotype most SNPs under very similar reaction conditions making it also highly flexible. The primer extension method is used in a number of assay formats. These formats use a wide range of detection techniques that include MALDI-TOF Mass spectrometry (see Sequenom) and ELISA-like methods (Rapley & Harbron 2004).
Generally, there are two main approaches which use the incorporation of either fluorescently labeled dideoxynucleotides (ddNTP) or fluorescently labeled deoxynucleotides (dNTP). With ddNTPs, probes hybridize to the target DNA immediately upstream of SNP nucleotide, and a single, ddNTP complementary to the SNP allele is added to the 3’ end of the probe (the missing 3'-hydroxyl in didioxynucleotide prevents further nucleotides from being added). Each ddNTP is labeled with a different fluorescent signal allowing for the detection of all four alleles in the same reaction. With dNTPs, allele-specific probes have 3’ bases which are complementary to each of the SNP alleles being interrogated. If the target DNA contains an allele complementary to the probe's 3’ base, the target DNA will completely hybridize to the probe, allowing DNA polymerase to extend from the 3’ end of the probe. This is detected by the incorporation of the fluorescently labeled dNTPs onto the end of the probe. If the target DNA does not contain an allele complementary to the probe's 3’ base, the target DNA will produce a mismatch at the 3’ end of the probe and DNA polymerase will not be able to extend from the 3' end of the probe. The benefit of the second approach is that several labeled dNTPs may get incorporated into the growing strand, allowing for increased signal. However, DNA polymerase in some rare cases, can extend from mismatched 3’ probes giving a false positive result (Rapley & Harbron 2004).
A different approach is used by Sequenom's iPLEX SNP genotyping method, which uses a MassARRAY mass spectrometer. Extension probes are designed in such a way that 40 different SNP assays can be amplified and analyzed in a PCR cocktail. The extension reaction uses ddNTPs as above, but the detection of the SNP allele is dependent on the actual mass of the extension product and not on a fluorescent molecule. This method is for low to medium high troughput, and is not intended for whole genome scanning.
The flexibility and specificity of primer extension make it amenable to high throughput analysis. Primer extension probes can be arrayed on slides allowing for many SNPs to be genotyped at once. Broadly referred to as arrayed primer extension (APEX), this technology has several benefits over methods based on differential hybridization of probes. Comparatively, APEX methods have greater discriminating power than methods using this differential hybridization, as it is often impossible to obtain the optimal hybridization conditions for the thousands of probes on DNA microarrays (usually this is addressed by having highly redundant probes). However, the same density of probes cannot be achieved in APEX methods, which translates into lower output per run (Rapley & Harbron 2004).
Illumina Incorporated's Infinium assay is an example of a whole-genome genotyping pipeline that is based on primer extension method. In the Infinium assay, over 100,000 SNPs can be genotyped. The assay uses hapten-labelled nucleotides in a primer extension reaction. The hapten label is recognized by anti-bodies, which in turn are coupled to a detectable signal (Gunderson et al. 2006).
APEX-2 is an arrayed primer extension genotyping method which is able to identify hundreds of SNPs or mutations in parallel using efficient homogeneous multiplex PCR (up to 640-plex) and four-color single-base extension on a microarray. The multiplex PCR requires two oligonucleotides per SNP/mutation generating amplicons that contain the tested base pair. The same oligonucleotides are used in the following step as immobilized single-base extension primers on a microarray (Krjutskov et al. 2008).
5’- nuclease
Taq DNA polymerase’s 5’-nuclease activity is used in the TaqMan assay for SNP genotyping. The TaqMan assay is performed concurrently with a PCR reaction and the results can be read in real-time as the PCR reaction proceeds (McGuigan & Ralston 2002). The assay requires forward and reverse PCR primers that will amplify a region that includes the SNP polymorphic site. Allele discrimination is achieved using FRET combined with one or two allele-specific probes that hybridize to the SNP polymorphic site. The probes will have a fluorophore linked to their 5’ end and a quencher molecule linked to their 3’ end. While the probe is intact, the quencher will remain in close proximity to the fluorophore, eliminating the fluorophore’s signal. During the PCR amplification step, if the allele-specific probe is perfectly complementary to the SNP allele, it will bind to the target DNA strand and then get degraded by 5’-nuclease activity of the Taq polymerase as it extends the DNA from the PCR primers. The degradation of the probe results in the separation of the fluorophore from the quencher molecule, generating a detectable signal. If the allele-specific probe is not perfectly complementary, it will have lower melting temperature and not bind as efficiently. This prevents the nuclease from acting on the probe (McGuigan & Ralston 2002).
Since the TaqMan assay is based on PCR, it is relatively simple to implement. The TaqMan assay can be multiplexed by combining the detection of up to seven SNPs in one reaction. However, since each SNP requires a distinct probe, the TaqMan assay is limited by the how close the SNPs can be situated. The scale of the assay can be drastically increased by performing many simultaneous reactions in microtitre plates. Generally, TaqMan is limited to applications that involve interrogating a small number of SNPs since optimal probes and reaction conditions must be designed for each SNP (Syvanen 2001).
Oligonucleotide ligase assay
DNA ligase catalyzes the ligation of the 3' end of a DNA fragment to the 5' end of a directly adjacent DNA fragment. This mechanism can be used to interrogate a SNP by hybridizing two probes directly over the SNP polymorphic site, whereby ligation can occur if the probes are identical to the target DNA. In the oligonucleotide ligase assay, two probes are designed; an allele-specific probe which hybridizes to the target DNA so that its 3' base is situated directly over the SNP nucleotide and a second probe that hybridizes the template upstream (downstream in the complementary strand) of the SNP polymorphic site providing a 5' end for the ligation reaction. If the allele-specific probe matches the target DNA, it will fully hybridize to the target DNA and ligation can occur. Ligation does not generally occur in the presence of a mismatched 3' base. Ligated or unligated products can be detected by gel electrophoresis, MALDI-TOF mass spectrometry or by capillary electrophoresis for large-scale applications (Rapley & Harbron 2004).
Other post-amplification methods based on physical properties of DNA
The characteristic DNA properties of melting temperature and single stranded conformation have been used in several applications to distinguish SNP alleles. These methods very often achieve high specificity but require highly optimized conditions to obtain the best possible results.
Single strand conformation polymorphism
Single-stranded DNA (ssDNA) folds into a tertiary structure. The conformation is sequence dependent and most single base pair mutations will alter the shape of the structure. When applied to a gel, the tertiary shape will determine the mobility of the ssDNA, providing a mechanism to differentiate between SNP alleles. This method first involves PCR amplification of the target DNA. The double-stranded PCR products are denatured using heat and formaldehyde to produce ssDNA. The ssDNA is applied to a non-denaturing electrophoresis gel and allowed to fold into a tertiary structure. Differences in DNA sequence will alter the tertiary conformation and be detected as a difference in the ssDNA strand mobility (Costabile et al. 2006). This method is widely used because it is technically simple, relatively inexpensive and uses commonly available equipment. However compared to other SNP genotyping methods, the sensitivity of this assay is lower. It has been found that the ssDNA conformation is highly dependent on temperature and it is not generally apparent what the ideal temperature is. Very often the assay will be carried out using several different temperatures. There is also a restriction on the length of fragment because the sensitivity drops when sequences longer than 400 bp are used (Costabile et al. 2006).
Temperature gradient gel electrophoresis
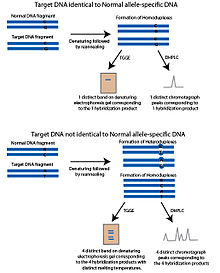
The temperature gradient gel electrophoresis (TGGE) or temperature gradient capillary electrophoresis (TGCE) method is based on the principle that partially denatured DNA is more restricted and travels slower in a porous material such as a gel. This property allows for the separation of DNA by melting temperature. To adapt these methods for SNP detection, two fragments are used; the target DNA which contain the SNP polymorphic site being interrogated and an allele-specific DNA sequence, referred to as the normal DNA fragment. The normal fragment is identical to the target DNA except potentially at the SNP polymorphic site, which is unknown in the target DNA. The fragments are denatured and then reannealed. If the target DNA has the same allele as the normal fragment, homoduplexes will form that will have the same melting temperature. When run on the gel with a temperature gradient, only one band will appear. If the target DNA has a distinct allele, four products will form following the reannealing step; homoduplexes consisting of target DNA, homoduplexes consisting of normal DNA and two heterduplexes of each strand of target DNA hybridized with the normal DNA strand. These four products will have distinct melting temperatures and will appear as four bands in the denaturing gel (Rapley & Harbron 2004).
Denaturing high performance liquid chromatography
Denaturing high performance liquid chromatography (DHPLC) uses reversed-phase HPLC to interrogate SNPs. The key to DHPLC is the solid phase which has differential affinity for single and double-stranded DNA. In DHPLC, DNA fragments are denatured by heating and then allowed to reanneal. The melting temperature of the reannealed DNA fragments determines the length of time they are retained in the column (Oefner & Underhill 1995). Using PCR, two fragments are generated; target DNA containing the SNP polymorphic site and an allele-specific DNA sequence, referred to as the normal DNA fragment. This normal fragment is identical to the target DNA except potentially at the SNP polymorphic site, which is unknown in the target DNA. The fragments are denatured and then allowed to gradually reanneal. The reannaled products are added to the DHPLC column. If the SNP allele in the target DNA matches the normal DNA fragment, only identical homoduplexes will form during the reannaling step. If the target DNA contains a different SNP allele than the normal DNA fragment, heteroduplexes of the target DNA and normal DNA containing a mismatched polymorphic site will form in addition to homoduplexes. The mismatched heteroduplexes will have a different melting temperature than the homoduplexes and will not be retained in the column as long. This generates a chromatograph pattern that is distinctive from the pattern that would be generated if the target DNA fragment and normal DNA fragments were identical. The eluted DNA is detected by UV absorption (Oefner & Underhill 1995).
DHPLC is easily automated as no labeling or purification of the DNA fragments is needed. The method is also relatively fast and has a high specificity. One major drawback of DHPLC is that the column temperature must be optimized for each target in order to achieve the right degree of denaturation (Rapley & Harbron 2004).
High-resolution melting of the entire amplicon
High Resolution Melting analysis is the simplest PCR-based method to understand. Basically, the same thermodynamic properties that allowed for the ingeniously crafted gel techniques to work apply here, and in real-time. A fluorimeter monitors the post-PCR denaturation of the entire dsDNA amplicon. You make primers specific to the site you want to amplify. You "paint" the amplicon with a double-strand specific dye, included in the PCR mix. The ds-specific dye integrates itself into the PCR product. in essence, the entire amplicon becomes a probe. This opens up new possibilities for discovery. Either you butt up the primers right up to the SNP in question (small amplicon genotyping, Liew, 2004) or amplify a larger region (100-400bp in length) for scanning purposes. For simple genotyping of an SNP, it is easier to just make the amplicon small to minimize the chances you mistake one SNP for another. The melting temperature (Tm) of the entire amplicon is determined and most homozygotes are sufficiently different (in the better instruments) in Tm to genotype. Heterozygotes are even easier to differentiate because they have heteroduplexes generated (refer to the gel-based explanations) which broadens the melt transition and usually gives two discernible peaks. Amplicon melting using a fluorescently-labeled primer has been described (Gundry et al., 2003) but is less practical than using ds-specific dyes due to the cost of the fluorogenic primer.
Scanning of larger amplicons is based on the same principles as outlined above. However, melting temperature and shape become informative. Numerous investigators have been able to successfully eliminate the majority of their sequencing through melt-based scanning. Many investigators have found scanning for mutations using high resolution melting as a viable and practical way to study entire genes.
Use of DNA mismatch-binding proteins
DNA mismatch-binding proteins can distinguish single nucleotide mismatches and thus facilitate differential analysis of SNPs. For example, MutS protein from Thermus aquaticus binds different single nucleotide mismatches with different affinities and can be used in capillary electrophoresis to differentiate all six sets of mismatches (Drabovich & Krylov 2006).
SNPlex
SNPlex is a proprietary genotyping platform sold by Applied Biosystems.
Sequencing
Next-generation sequencing technologies such as pyrosequencing sequence less than 250 bases in a read which limits their ability to sequence whole genomes. However, their ability to generate results in real-time and their potential to be massively scaled up makes them a viable option for sequencing small regions to perform SNP genotyping. Compared to other SNP genotyping methods, sequencing is in particular, suited to identifying multiple SNPs in a small region, such as the highly polymorphic Major Histocompatibility Complex region of the genome (Rapley & Harbron 2004).
References
- Abravaya K., Huff J., Marshall R., Merchant B., Mullen C., Schneider G., and Robinson J. (2003) Molecular beacons as diagnostic tools: technology and applications. Clin Chem Lab Med. 41:468-474.
- Costabile M., Quach A., Ferrante A. (2006) Molecular approaches in the diagnosis of primary immunodeficiency diseases. Hum Mutat. 27(12):1163-73.
- Drabovich A.P., Krylov S.N. (2006). Identification of base pairs in single-nucleotide polymorphisms by MutS protein-mediated capillary electrophoresis. Analytical chemistry. 78 (6): 2035-2038.
- Gundry C.N., Vandersteen J.G., Reed G.H., Pryor R.J., Chen J., Wittwer C.T. (2003) Amplicon Melting Analysis with Labeled Primers: A Closed-Tube Method for Differentiating Homozygotes and Heterozygotes. Clin Chem. 49(3):396-406.
- Gunderson K.L., Steemers F.J., Ren H., et al.. (2006) Whole-genome genotyping. Methods Enzymol. 410:359-76
- Howell W., Jobs M., Gyllensten U., Brookes A. (1999) Dynamic allele-specific hybridization. A new method for scoring single nucleotide polymorphisms. Nat Biotechnol. 17(1):87-8.
- Liew M., Pryor R., Palais R., Meadows C., Erali M., Lyon E., Wittwer C. (2004) Genotyping of Single-Nucleotide Polymorphisms by High-Resolution Melting of Small Amplicons. Clin Chem. 50(7):1156-1164.
- McGuigan F.E., Ralston S.H. (2002) Single nucleotide polymorphism detection: allelic discrimination using TaqMan. Psychiatr Genet. 12(3):133-6.
- Oefner, P.J., Underhill, P.A. (1995) Comparative DNA sequencing by denaturing high-performance liquid chromatography (DHPLC). Am J Hum Genet. 57, A266.
- Olivier M. (2005) The Invader assay for SNP genotyping. Mutat Res. 573(1-2):103-10.
- Rao K.V., Stevens P.W., Hall J.G., et al. (2003) Genotyping single nucleotide polymorphisms directly from genomic DNA by invasive cleavage reaction on microspheres. Nucleic Acids Res. 31(11):e66.
- Rapley R., Harbron S. (Eds.) (2004) Molecular Analysis and Genome Discovery. Chichester. John Wiley & Sons Ltd.
- Syvanen A.C. (2001) Accessing genetic variation: genotyping single nucleotide polymorphisms. Nat Rev Genet. 2(12):930-42.
- Affymetrix (2007) Genome-Wide Human SNP Array 5.0. [online] Address: http://www.affymetrix.com/products/arrays/specific/genome_wide/genome_wide_snp_5.affx. Retrieved on February 27, 2007.
External links
- International HapMap Project
- Delaware Valley Personalized Medicine Project Uses SNPs to help make medicine personal
- Genotyping by Oligonucleotide Ligation Assay (OLA)
Categories:




Wikimedia Foundation. 2010.