- Overlap extension polymerase chain reaction
-
-
- This page assumes familiarity with the terms and components used in polymerase chain reaction (PCR) process.
The overlap extension polymerase chain reaction (or OE-PCR) is a variant of PCR. It is also referred to as Splicing by overlap extension / Splicing by overhang extension (SOE) PCR. It is used to insert specific mutations at specific points in a sequence or to splice smaller DNA fragments into a larger polynucleotide.[1]Splicing of DNA Molecules
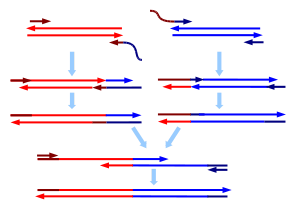 This image shows how OE-PCR might be utilized to splice two DNA sequences (red and blue). The arrows represent the 3' ends
This image shows how OE-PCR might be utilized to splice two DNA sequences (red and blue). The arrows represent the 3' ends
As in most PCR reaction, two primers -one for each end- are used per sequence. To splice two DNA molecules, special primers are used at the ends that are to be joined. For each molecule, the primer at the end to be joined is constructed such that it has a 5' overhang complementary to the end of the other molecule. Following annealing when replication occurs, the DNA is extended by a new sequence that is complementary to the molecule it is to be joined to. Once both DNA molecules are extended in such a manner, they are mixed and a PCR is carried out with only the primers for the far ends. The overlapping complementary sequences introduced will serve as primers and the two sequences will be fused. This method has an advantage over other gene splicing techniques in not requiring restriction sites.
To get higher yields, some primers are used in excess as in asymmetric PCR.
Introduction of Mutations
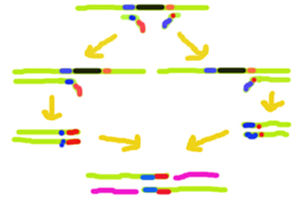 This image shows how OE-PCR might be utilized to delete a sequence from a DNA strand
This image shows how OE-PCR might be utilized to delete a sequence from a DNA strandTo insert a mutation into a DNA sequence, a specific primer is designed. The primer may contain a single substitution or contain a new sequence at its 5' end. If a deletion is required, a sequence that is 5' of the deletion is added, because the 3' end of the primer must have complementarity to the template strand so that the primer can sufficiently anneal to the template DNA.
Following annealing of the primer to the template, DNA replication proceeds to the end of the template. The duplex is denatured and the second primer anneals to the newly formed DNA strand, containing sequence from the first primer. Replication proceeds to produce a strand of the required sequence, containing the mutation.
The duplex is denatured again and the first primer can now bind to the latest DNA strand. The replication reaction continues to produce a fully dimerised DNA fragment. After further PCR cycles, to amplify the DNA, the sample can be separated by agarose gel electrophoresis, followed by electroelution for collection.
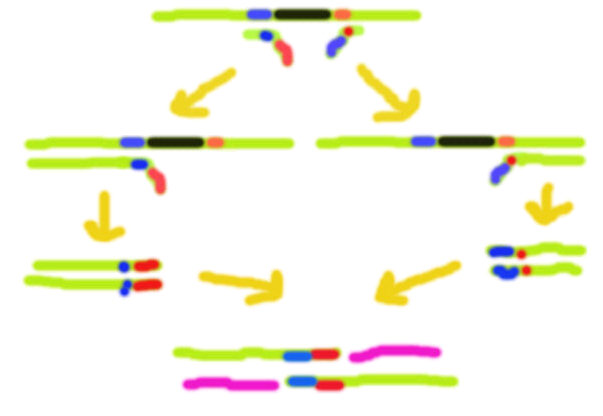
Efficiently generating oligonucleotides beyond ~110 nucleotides in length is very difficult, so to insert a mutation further into a sequence than a 110 nt primer will allow, it is necessary to employ overlap extension PCR. In OE-PCR the sequence being modified is used to make two modified strands with the mutation at opposite ends, using the technique described above. After mixing and denaturation, the strands are allowed to anneal to produce three different combinations as detailed in the diagram. Only the duplex without overlap at the 5' end will allow extension by DNA polymerase in 3' to 5' direction.
Following separation, the eluted fragments of appropriate size are subject to normal PCR, using the outermost primers used in the initial, mutagenic PCR reactions.
References
- ^ Higuchi R, Krummel B, Saiki R (1988). "A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions". Nucleic Acids Res 16 (15): 7351–67. doi:10.1093/nar/16.15.7351. PMC 338413. PMID 3045756. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=338413.
Polymerase chain reaction techniques Real-time polymerase chain reaction (QRT-PCR) | Reverse transcription polymerase chain reaction (RT-PCR) | Inverse polymerase chain reaction | Nested polymerase chain reaction | Touchdown polymerase chain reaction | Overlap extension polymerase chain reaction | Multiplex polymerase chain reaction | Multiplex ligation-dependent probe amplification
Categories:- Molecular biology
- Laboratory techniques
- Polymerase chain reaction
- Genetic engineering
- Molecular biology techniques
-

Wikimedia Foundation. 2010.