- 1,3-Dipolar cycloaddition
-
The 1,3-dipolar cycloaddition, also known as the Huisgen cycloaddition or Huisgen reaction, [1][2] is an organic chemical reaction belonging to the larger class of concerted, pericyclic cycloadditions. It is the reaction between a 1,3-dipole and a dipolarophile, most of which are substituted alkenes, to form a five-membered ring. Rolf Huisgen first saw the prospects of varying the 1,3-dipole and its high value for synthesis of 5-membered heterocycles.
Contents
Proof of mechanism
Although this was once hotly contested, the possibility of a diradical intermediate, defended by Firestone at the time, has largely been disproved by the work of Huisgen. It is now accepted that the mechanism is a concerted process.[3]
Proof of the concerted mechanism:
- Increasing carbonyl groups on the 1,3-dipole decreases reactivity by ground state stabilization.
- Solvent effects are minor so the reaction occurs through the concerted mechanism which lacks a zwitterionic intermediate. If the reaction occurred through a charge-separated intermediate, an increase in reaction rate with solvent polarity would be expected.
- The reaction proceeds with cis-addition as two bonds are formed simultaneously so there is no intermediate that can rotate to scramble the product stereochemistry.
Countless additional calculations and experiments have been performed in addition to verify the concerted mechanism.
Reactivity
Concerted processes such as the 1,3-cycloaddition require a highly ordered transition state (high negative entropic energy) and only moderate enthalpy requirements. Using competition reaction experiments, relative rates of addition for different cycloaddition reactions have been found to offer general findings on factors in reactivity.
- Conjugation, especially with aromatic groups, increases the rate of reaction by stabilization of the transition state. During the transition, the two sigma bonds are being formed at different rates, which may generate partial charges in the transition state that can be stabilized by charge distribution into conjugated substituents.
- More polarizable dipolarophiles are more reactive because diffuse electron clouds are better suited to initiate the flow of electrons.
- Dipolarophiles with high angular strain are more reactive due to increased energy of the ground state.
- Increased steric hindrance in the transition state as a result of unhindered reactants dramatically lowers the reaction rate.
- Hetero-dipolarophiles add more slowly, if at all, compared to C,C-diapolarophiles due to a lower gain in sigma bond energy to offset the loss of a pi bond during the transition state.
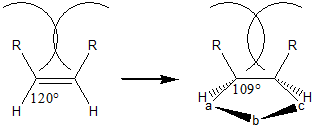
- Isomerism of the dipolarophile affects reaction rate due to sterics. trans-isomers are more reactive (trans-stilbene will add diphenyl(nitrile imine) 27 times faster than cis-stilbene) because during the reaction, the 120° bond angle shrinks to 109°, bringing eclipsing cis-substituents towards each other for increased steric clash.
Product orientation
Although there are general guidelines for orientation of products, the actual selectivity in these cycloaddition reactions is varied. For example, in azide-alkyne coupling reactions without Cu(I) catalyst, the products are generated in a 1:1 ratio of 1,4 and 1,5 products [4].
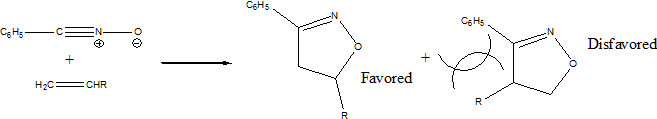
In reactions with hetero-dipolarophiles, the direction of addition is based on the maximization of gains in sigma bond energy during reaction. However, there are several exceptions that include multistep reaction pathways.
When the dipolarophiles are alkenes or alkynes, the dominant force in dictating direction of addition are substituents effects, primarily steric. [5]
Molecular Orbitals
The reaction goes through a symmetry-allowed Huckel aromatic molecular orbital in the transition state. The allyl anion-type orbital of the dipole is involved in the reaction, where the terminal centers are both nucleophilic and electrophilic, depending on the resonance structures.
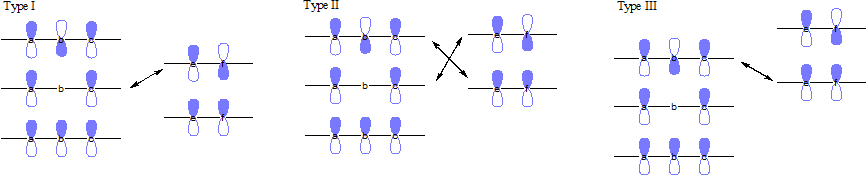
In an FMO consideration of 1,3-cycloadditions, both combinations of HOMO-LUMO considerations contribution to the interaction energies. These reactions can be categorized into three major types based on the conjugated substituents.
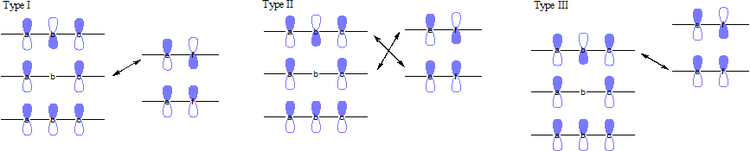
- Type I: Electron-withdrawing groups favor reaction by stabilizing the dipolarophiles LUMO and decreasing the HOMO-LUMO gap. Electron-donating groups will deactivate dipolarophiles.
- Type II: Electron-withdrawing groups lower the orbital energies of the dipolarophiles. Electron-releasing substituents raise the dipolarophiles orbital energies. In both cases, one HOMO-LUMO gap is decreased more than the other is increased, so the overall result is an increase in reaction rate.
- Type III: Electron-donating groups increase the dipolarophiles HOMO to better interact with the 1,3-dipole LUMO. Electron-withdrawing groups will deactivate the dipolarophiles.[6]
Classes of 1,3-dipoles
Huisgen and others investigated a range of 1,3-dipoles and dipolarophiles. These included:
- Azides and alkynes react in the Azide alkyne Huisgen cycloaddition.
- Diazo compounds are 1,3-dipoles in the Diazoalkane 1,3-dipolar cycloaddition.
- Nitrones react with alkenes to form isoxazolidines.
- Nitrile oxides react with alkenes to form isoxazolines.
- Ozonolysis begins with a 1,3-dipolar cycloaddition of ozone. This is followed by a retro-1,3-dipolar cycloaddition and then a 1,3-dipolar cycloaddition of the fragments.
- Fullerenes and nanotubes can undergo a 1,3-dipolar cycloaddition with an azomethine ylide in the Prato reaction.
References
- ^ Huisgen, Rolf (November 1963). "Kinetics and Mechanism of 1,3-Dipolar Cycloadditions" (abstract). Angewandte Chemie International Edition 2 (11): 633–645. doi:10.1002/anie.196306331. http://www3.interscience.wiley.com/cgi-bin/abstract/106572717/ABSTRACT.
- ^ Huisgen, Rolf (October 1963). "1,3-Dipolar Cycloadditions. Past and Future" (abstract). Angewandte Chemie International Edition 2 (10): 565–598. doi:10.1002/anie.196305651. http://www3.interscience.wiley.com/cgi-bin/abstract/106572678/ABSTRACT.
- ^ Huisgen, R. (1976). "The Concerted Nature of 1,3-Dipolar Cycoadditions and the Question of Diradical Intermediates". The Journal of Organic Chemistry 41 (3): 403-419. doi:10.1021/jo00865a001.
- ^ Vsevolod V. Rostovtsev, Luke G. Green, Valery V. Fokin, K. Barry Sharpless (2002). "A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective Ligation of Azides and Terminal Alkynes". Angewandte Chemie International Edition 41 (14): 2596–22599. doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4.
- ^ Huisgen, Rolf (November 1963). "Kinetics and Mechanism of 1,3-Dipolar Cycloadditions" (abstract). Angewandte Chemie International Edition 2 (11): 633–645. doi:10.1002/anie.196306331. http://www3.interscience.wiley.com/cgi-bin/abstract/106572717/ABSTRACT.
- ^ Huisgen, R. (1976). "The Concerted Nature of 1,3-Dipolar Cycoadditions and the Question of Diradical Intermediates". The Journal of Organic Chemistry 41 (3): 403-419. doi:10.1021/jo00865a001.
External links
- Click Chemicals. A new site featuring in depth discussion, faq's and links to key papers surround 1,3 dipolar cycloadditions
Categories:- Cycloadditions

Wikimedia Foundation. 2010.