- Bioavailability
-
In pharmacology, bioavailability (BA) is a subcategory of absorption and is used to describe the fraction of an administered dose of unchanged drug that reaches the systemic circulation, one of the principal pharmacokinetic properties of drugs. By definition, when a medication is administered intravenously, its bioavailability is 100 %.[1] However, when a medication is administered via other routes (such as orally), its bioavailability generallyTH[›] decreases (due to incomplete absorption and first-pass metabolism) or may vary from patient to patient. Bioavailability is one of the essential tools in pharmacokinetics, as bioavailability must be considered when calculating dosages for non-intravenous routes of administration.
For dietary supplements, herbs and other nutrients in which the route of administration is nearly always oral, bioavailability generally designates simply the quantity or fraction of the ingested dose that is absorbed.[2]
Bioavailability is defined slightly differently for drugs as opposed to dietary supplements primarily due to the method of administration and Food and Drug Administration regulations.
Bioaccessibility is a concept related to bioavailability in the context of biodegradation and environmental pollution. A molecule (often a persistent organic pollutant) is said to be bioavailable when "[it] is available to cross an organism’s cellular membrane from the environment, if the organism has access to the chemical." [3]
Definitions
In pharmacology
In pharmacology, bioavailability is a measurement of the extent to which a drug reaches the systemic circulation.[4] It is denoted by the letter f (or, if expressed in percent, by F).
In nutritional sciences
In nutritional sciences, which covers the intake of nutrients and non-drug dietary ingredients, the concept of bioavailability lacks the well-defined standards associated with the pharmaceutical industry. The pharmacological definition cannot apply to these substances because utilization and absorption is a function of the nutritional status and physiological state of the subject,[5] resulting in even greater differences from individual to individual (inter-individual variation). Therefore, bioavailability for dietary supplements can be defined as the proportion of the substance capable of being absorbed and available for use or storage.[6]
In both pharmacology and nutrition sciences, bioavailability is measured by calculating the area under curve (AUC) of the drug concentration time profile.
In environmental sciences
Bioavailability is commonly a limiting factor in the production of crops (due to solubility limitation or adsorption of plant nutrients to soil colloids) and in the removal of toxic substances from the food chain by microorganisms (due to sorption to or partitioning of otherwise degradable substances into inaccessible phases in the environment). A noteworthy example for agriculture is plant phosphorus deficiency induced by precipitation with iron and aluminum phosphates at low soil pH and precipitation with calcium phosphates at high soil pH.[7] Toxic materials in soil, such as lead from sloughed paint may be rendered unavailable to animals ingesting contaminated soil by supplying phosphorus fertilizers in excess.[8] Organic pollutants such as solvents or pesticides may be rendered unavailable to microorganisms and thus persist in the environment when they are adsorbed to soil minerals[9] or partition into hydrophobic organic matter.[10]
Absolute bioavailability
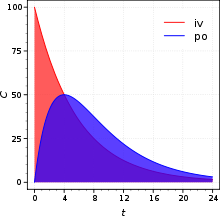 Bioavailability is a ratio of areas under the curves
Bioavailability is a ratio of areas under the curves
Absolute bioavailability compares the bioavailability of the active drug in systemic circulation following non-intravenous administration (i.e., after oral, rectal, transdermal, subcutaneous, or sublingual administration), with the bioavailability of the same drug following intravenous administration. It is the fraction of the drug absorbed through non-intravenous administration compared with the corresponding intravenous administration of the same drug. The comparison must be dose normalized (e.g. account for different doses or varying weights of the subjects); consequently, the amount absorbed is corrected by dividing the corresponding dose administered.
In pharmacology, in order to determine absolute bioavailability of a drug, a pharmacokinetic study must be done to obtain a plasma drug concentration vs time plot for the drug after both intravenous (iv) and extravascular (non-intravenous, i.e., oral) administration. The absolute bioavailability is the dose-corrected area under curve (AUC) non-intravenous divided by AUC intravenous. For example, the formula for calculating F for a drug administered by the oral route (po) is given below.
Therefore, a drug given by the intravenous route will have an absolute bioavailability of 100 % (f=1) while drugs given by other routes usually have an absolute bioavailability of less than one. If we compare the two different dosage forms having same active ingredients and compare the two drug bioavailability is called comparative bioavailability. Although knowing the true extent of systemic absorption (referred to as absolute bioavailability) is clearly useful, in practice it is not determined as frequently as one may think. The reason for this is that its assessment requires an intravenous reference, that is, a route of administration that guarantees that all of the administered drug reaches the systemic circulation. Such studies come at considerable cost, not least of which is the necessity to conduct preclinical toxicity tests to ensure adequate safety, as well as there being potential problems due to solubility limitations. These limitations may be overcome however, by administering a very low dose (typically a few micrograms) of an isotopically labelled drug concomitantly with a therapeutic non-labelled oral dose. Providing the isotopically-labelled intravenous dose is sufficiently low so as not to perturb the systemic drug concentrations achieved from the absorbed oral dose, then the intravenous and oral pharmacokinetics can be deconvoluted by virtue of the their different isotopic constitution and thereby determine the oral and intravenous pharmacokinetics from the same dose administration. This technique eliminates pharmacokinetic issues on non-equivalent clearance as well as enabling the intravenous dose to be administered with a minimum of toxicology and formulation. The technique was first applied using stable-isotopes such as C-13 and mass-spectroscopy to distinguish the isotopes by mass difference. More recently, C-14 labelled drugs are administered intravenously and accelerator mass spectrometry (AMS) used to measure the isotopically labelled drug along with mass spectrometry for the unlabelled drug.[11]
There is no regulatory requirement to define the intravenous pharmacokinetics or absolute bioavailability however regulatory authorities do sometimes ask for absolute bioavailbility information of the extravascular route in cases in which the bioavailability is apparently low or variable and there is a proven relationship between the pharmacodynamics and the pharmacokinetics at therapeutic doses. In all such cases, to conduct an absolute bioavailability study requires that the drug be given intravenously.[12]
Intravenous administration of a developmental drug can provide valuable information on the fundamental pharmacokinetic parameters of volume of distribution (V) and clearance (CL).[12]
Relative bioavailability and bioequivalence
In pharmacology, relative bioavailability measures the bioavailability (estimated as the AUC) of a formulation (A) of a certain drug when compared with another formulation (B) of the same drug, usually an established standard, or through administration via a different route. When the standard consists of intravenously administered drug, this is known as absolute bioavailability (see above).
Relative bioavailability is one of the measures used to assess bioequivalence (BE) between two drug products. For FDA approval a generic manufacturer must demonstrate that the 90 % confidence interval for the ratio of the mean responses (usually of AUC and the maximum concentration, Cmax) of its product to that of the "Brand Name drug"OB[›] is within the limits of 80 % to 125 %. While AUC refers to the extent of bioavailability, Cmax refers to the rate of bioavailability. When Tmax is given, it refers to the time it takes for a drug to reach Cmax.
While the mechanisms by which a formulation affects bioavailability and bioequivalence have been extensively studied in drugs, formulation factors that influence bioavailability and bioequivalence in nutritional supplements are largely unknown.[13] As a result, in nutritional sciences, relative bioavailability or bioequivalence is the most common measure of bioavailability, comparing the bioavailability of one formulation of the same dietary ingredient to another.
Factors influencing bioavailability
The absolute bioavailability of a drug, when administered by an extravascular route, is usually less than one (i.e. F <100 %). Various physiological factors reduce the availability of drugs prior to their entry into the systemic circulation. Whether a drug is taken with or without food will also affect absorption, other drugs taken concurrently may alter absorption and first-pass metabolism, intestinal motility alters the dissolution of the drug and may affect the degree of chemical degradation of the drug by intestinal microflora. Disease states affecting liver metabolism or gastrointestinal function will also have an effect.
Other factors may include, but are not limited to:
- Physical properties of the drug (hydrophobicity, pKa, solubility)
- The drug formulation (immediate release, excipients used, manufacturing methods, modified release – delayed release, extended release, sustained release, etc.)
- Whether the formulation is administered in a fed or fasted state
- Gastric emptying rate
- Circadian differences
- Interactions with other drugs/foods:
- Interactions with other drugs (e.g. antacids, alcohol, nicotine)
- Interactions with other foods (e.g. grapefruit juice, pomello, cranberry juice, brassica vegetables)
- Transporters: Substrate of efflux transporters (e.g. P-glycoprotein)
- Health of the GI tract
- Enzyme induction/inhibition by other drugs/foods:
- Individual variation in metabolic differences
- Age: In general, drugs are metabolized more slowly in fetal, neonatal, and geriatric populations
- Phenotypic differences, enterohepatic circulation, diet, gender
- Disease state
- e.g. hepatic insufficiency, poor renal function
Each of these factors may vary from patient to patient (inter-individual variation), and indeed in the same patient over time (intra-individual variation). In clinical trials, inter-individual variation is a critical measurement used to assess the bioavailability differences from patient to patient in order to ensure predictable dosing.
Bioavailability of drugs versus dietary supplements
In comparison to drugs, there are significant differences in dietary supplements that impact the evaluation of their bioavailability. These differences include the following: the fact that nutritional supplements provide benefits that are variable and often qualitative in nature; the measurement of nutrient absorption lacks the precision; nutritional supplements are consumed for prevention and well-being; nutritional supplements do not exhibit characteristic dose-response curves; and dosing intervals of nutritional supplements, therefore, are not critical in contrast to drug therapy.[6]
In addition, the lack of defined methodology and regulations surrounding the consumption of dietary supplements hinders the application of bioavailability measures in comparison to drugs. In clinical trials with dietary supplements, bioavailability primarily focuses on statistical descriptions of mean or average AUC differences between treatment groups, while often failing to compare or discuss their standard deviations or inter-individual variation. This failure leaves open the question of whether or not an individual in a group is likely to experience the benefits described by the mean-difference comparisons. Further, even if this issue were discussed, it would be difficult to communicate meaning of these inter-subject variances to consumers and/or their physicians.
Nutritional science: reliable and universal bioavailability
One way to resolve this problem is to define "reliable bioavailability" as positive bioavailability results (an absorption meeting a predefined criteria) that include 84 % of the trial subjects and "universal bioavailability" as those that include 98 % of the trial subjects. This reliable-universal framework would improve communications with physicians and consumers such that, if it were included on products labels for example, make educated choices as to the benefits of a formulation for them directly. In addition, the reliable-universal framework is similar to the construction of confidence intervals, which statisticians have long offered as one potential solution for dealing with small samples, violations of statistical assumptions or large standard deviations.[14]
Notes
^ TH: One of the few exceptions where a drug shows F of >100 % is theophylline. If administered as an oral solution F is 111 %, since the drug is completely absorbed and first-past metabolism in the lung after iv administration is bypassed.[15]
^ OB: Reference listed drug products (i.e. innovator's) as well as generic drug products which have been approved based on an Abbreviated New Drug Application are given in FDA's "Orange Book".See also
- ADME-Tox
- Lipinski's Rule of 5
- Biopharmaceutics Classification System
- Caco-2
Footnotes
- ^ Griffin, J.P. The Textbook of Pharmaceutical Medicine (6th Ed.). New Jersey: BMJ Books. ISBN 9781405180351
- ^ Factors Influencing the Measurement of Bioavailability, Taking Calcium as a Model. Robert P. Heaney. J. Nutr. (2001) 131:1344S-1348S
- ^ Semple KT, Doick KJ, Jones KC, Burauel P, Craven A, Harms H (2004). "Defining bioavailability and bioaccessibility of contaminated soil and sediment is complicated". Environ. Sci. Technol. 38 (12): 228A–231A. doi:10.1021/es040548w. PMID 15260315. http://pubs.acs.org/doi/abs/10.1021/es040548w.
- ^ Shargel, L.; Yu, A.B. (1999). Applied biopharmaceutics & pharmacokinetics (4th ed.). New York: McGraw-Hill. ISBN 0-8385-0278-4
- ^ Heaney RP (2001). "Factors Influencing the Measurement of Bioavailability, Taking Calcium as a Model". J. Nutr. 131 (4 Suppl): 1344S-1348S. PMID 11285351.
- ^ a b Srinivasan VS (2001). "Bioavailability of Nutrients: A Practical Approach to In Vitro Demonstration of the Availability of Nutrients in Multivitamin-Mineral Combination Products". J. Nutr. 131 (4 Suppl): 1349S-1350S. PMID 11285352.
- ^ Hinsinger, P. 2001. Bioavailability of soil inorganic P in the rhizosphere as affected by root-induced chemical changes: a review. Plant and Soil 237: 173–195.
- ^ Ma Q.Y., Traina S.J., Logan T.J. 1993. In situ lead immobilization by apatite. Environ Sci Technol 27:1803–1810.
- ^ O'Loughlin, E. J, S. J. Traina, and G. K. Sims. 2000. Effects of sorption on the biodegradation of 2-methylpyridine in aqueous suspensions of reference clay minerals. Environ. Toxicol. and Chem. 19:2168-2174.
- ^ Sims, G. K. and A.M. Cupples. 1999. Factors controlling degradation of pesticides in soil. Pesticide Science 55:598-601.
- ^ Graham Lappin, Malcolm Rowland, R. Colin Garner. The use of Isotopes in the Determination of Absolute Bioavailability of Drugs in Humans. Expert Opin. Drug Metab. Toxicol. (2006) 2(3)
- ^ a b Graham Lappin, Lloyd Stevens. Biomedical accelerator mass spectrometry: recent applications in metabolism and pharmacokinetics. Expert Opin. Drug Metab. Toxicol. (2008) 4(8):1021-1033
- ^ Hoag SW, Hussain AS (2001). "The Impact of Formulation on Bioavailability: Summary of Workshop Discussion.". J. Nutr. 131 (4 Suppl): 1389S-1391S. PMID 11285360.
- ^ Kagan D, Madhavi D, Bank G, Lachlan K (2010). ""Universal" and "Reliable" Bioavailability Claims: Criteria That May Increase Physician Confidence in Nutritional Supplements.". Natural Medicine Journal. 2 (1): 1–5.[1]
- ^ Schuppan, D.; K. H. Molz, A. H. Staib, N. Rietbrock (1981). "Bioavailability of theophylline from a sustained-release aminophylline formulation (Euphyllin retard tablets)–plasma levels after single and multiple oral doses". Int J Clin Pharmacol Ther Toxicol 19 (5): 223–227. PMID 7251238. "Absorption of theophylline from solution was rapid and complete. Bioavailability amounted to 111 ± 16% compared to i. v. administration."
References
- Malcolm Rowland; Thomas N. Tozer (2010). Clinical Pharmacokinetics and Pharmacodynamics: Concepts and Applications (4 ed.). Philadelphia, PA: Lippincott Williams & Wilkins. ISBN 9780781750097.
- Peter G. Welling; Francis L. S. Tse; Shrikant V. Dighe (1991). Pharmaceutical Bioequivalence. Drugs and the Pharmaceutical Sciences. 48. New York, NY: Marcel Dekker. ISBN 9780824784843.
- Dieter Hauschke; Volker Steinijans; Iris Pigeot (2007). "Metrics to characterize concentration-time profiles in single- and multiple-dose bioequivalence studies". Bioequivalence Studies in Drug Development: Methods and Applications. Statistics in Practice. Chichester, UK: John Wiley and Sons. pp. 17–36. ISBN 9780470094754. http://books.google.com/books?id=mVOOyubcXqwC&pg=PA17. Retrieved 21 April 2011.
- Shein-Chung Chow; Jen-pei Liu (15 October 2008). Design and Analysis of Bioavailability and Bioequivalence Studies. Biostatistics Series. 27 (3 ed.). Boca Raton, FL: CRC Press. ISBN 9781584886686.
External links
Topics in Medicinal Chemistry ADME · Bioavailability · Chemogenomics · Drug design · Drug discovery · Enzyme inhibitor · Ligand efficiency · Mechanism of action · New chemical entity · Pharmacodynamics · Pharmacokinetics · Pharmacology · Pharmacophore · Quantitative structure-activity relationshipMedication > Pharmacology Pharmacokinetics ADME: Absorption • Distribution • Metabolism • Excretion (Clearance)
Loading dose • Volume of distribution (Initial) • Rate of infusion
Compartment • Bioequivalence • Bioavailability
Onset of action • Biological half-life • Plasma protein binding
Therapeutic index (LD50/ED50)Pharmacodynamics Agonism and antagonism Agonist: Inverse agonist • Irreversible agonist • Partial agonist • Superagonist • Physiological agonist
Antagonist: Competitive antagonist • Irreversible antagonist • Physiological antagonist
Other: Binding • Affinity • Binding selectivity • Functional selectivityOther Related fields/subfields Categories:



Wikimedia Foundation. 2010.