- Metal L-edge
-
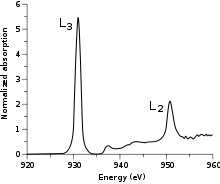 Figure 1: L3- and L2-edges of [CuCl4]2-.
Figure 1: L3- and L2-edges of [CuCl4]2-.
 Figure 2: L-edge spectral components.
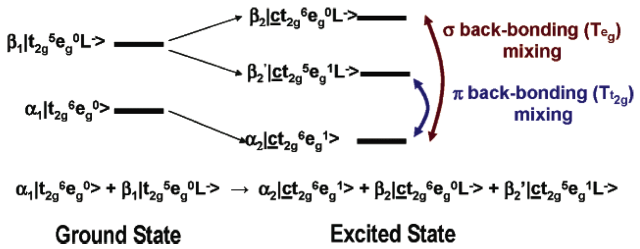
Figure 2: L-edge spectral components.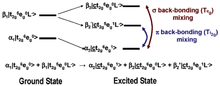 Figure 3: Configurations involved in the ground and excited states and the mechanisms by which the intensity of the L-edge features can mix.
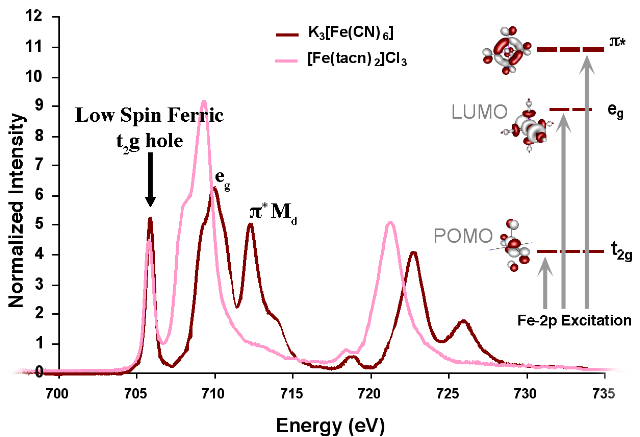
Figure 3: Configurations involved in the ground and excited states and the mechanisms by which the intensity of the L-edge features can mix.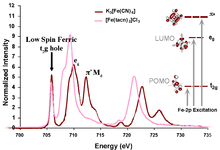 Figure 4: Comparison of the Fe L-edges of low-spin K3[Fe(CN)6] and [Fe(tacn)2]Cl3. Tacn is a σ only donor, meaning no backbonding and only two main L-edge features. K3[Fe(CN)6] has significant backbonding, as is shown by the third transition to higher energy in the L-edge spectrum.
Figure 4: Comparison of the Fe L-edges of low-spin K3[Fe(CN)6] and [Fe(tacn)2]Cl3. Tacn is a σ only donor, meaning no backbonding and only two main L-edge features. K3[Fe(CN)6] has significant backbonding, as is shown by the third transition to higher energy in the L-edge spectrum.
Metal L-edge XAS is an experimental technique that involves the excitation of a metal 2p electron to unfilled metal d orbitals (e.g. 3d for first-row transition metals). According to the selection rules, the transition is formally electric-dipole allowed, which not only makes it more intense than an electric-dipole forbidden metal K pre-edge[1] (1s → 3d transition), but also makes it more feature-rich as the lower required energy (~400-1000 eV scandium to copper) results in a higher-resolution experiment.[2] In the simplest case, that of a cupric (CuII) complex, the 2p → 3d transition produces a 2p53d10 final state. The 2p5 core hole created in the transition has an orbital angular momentum L=1 which then couples to the spin angular momentum S=1/2 to produce J = 3/2 and J=1/2 final states. These states are directly observable in the L-edge spectrum as the two main peaks (Figure 1). The peak at lower energy (~930 eV) has the greatest intensity and is called the L3-edge while the peak at higher energy (~950 eV) has less intensity and is called the L2-edge.
As we move left across the periodic table (e.g. from copper to iron), we create additional holes in the metal 3d orbitals. For example, a low-spin ferric (FeIII) system in an octahedral environment has a ground state of (t2g)5(eg)0 resulting in transitions to the t2g (dπ) and eg (dσ) sets. Therefore, there are two possible final states: t2g6eg0 or t2g5eg1(Figure 2a). Since the ground-state metal configuration has one hole in the eg orbital set and four holes in the t2g orbital set, an intensity ratio of 1:4 might be expected (Figure 2b). However, this model does not take into account covalent bonding and, indeed, an intensity ratio of 1:4 is not observed in the spectrum. In the case of iron, the d6 excited state will further split in energy due to d-d electron repulsion (Figure 2c). This splitting is given by the right-hand (high-field) side of the d6 Tanabe-Sugano diagram and can be mapped onto a theoretical simulation of a L-edge spectrum (Figure 2d). Other factors such as p-d electron repulsion and spin-orbit coupling of the 2p and 3d electrons must also be considered to fully simulate the data. For a ferric system, all of these effects result in 252 initial states and 1260 possible final states that together will comprise the final L-edge spectrum (Figure 2e). Despite all of these possible states, it has been established that in a low-spin ferric system, the lowest energy peak is due to a transition to the t2g hole and the more intense and higher energy (~3.5 eV) peak is to that of the unoccupied eg orbitals.[3]
In most systems, bonding between a ligand and a metal atom can be thought of in terms of metal-ligand covalent bond where the occupied ligand orbitals donates some electron density to the metal. This is commonly known as ligand to metal charge transfer or LMCT. In some cases, low-lying unoccupied ligand orbitals (π*) can receive back-donation (or backbonding) from the occupied metal orbitals. This has the opposite effect on the system, resulting in metal to ligand charge transfer, MLCT, and commonly appears as an additional L-edge spectral feature. An example of this feature occurs in low-spin ferric [Fe(CN)6]3- since CN- is a ligand that can have back-bonding. While back-bonding is important in the initial state, it would only warrant a small feature in the L-edge spectrum. In fact, it is in the final state where the back bonding π* orbitals are allowed to mix with the very intense eg transition, thus borrowing intensity and resulting in the final dramatic three peak spectrum (Figure 3 and Figure 4).[4]XAS, as well as other spectroscopies, look at the excited state to infer information about the ground state. Thus, spectra of this nature contain many features that have to be accounted for and understood. To make a quantitative assignment, L-edge data is fit using a valence bond configuration interaction (VBCI) model where LMCT and MLCT are applied as needed to successfully simulate the observed spectral features.[3] These simulations are then further compared to density functional theory (DFT) calculations to arrive at a final interpretation of the data and an accurate description of the electronic structure of the complex (Figure 4).
In the case of iron L-edge, the excited state mixing of the metal eg orbitals into the ligand π* make this method a direct and very sensitive probe of backbonding.[4]
References
- ^ T. E. Westre, P. Kennepohl, J. G. DeWitt, B. Hedman, K. O. Hodgson, E. I. Solomon. "A Multiplet Analysis of Fe K-Edge 1s → 3d Pre-Edge Features of Iron Complexes" J. Am. Chem. Soc. 1997, 119, pp. 6297-6314.
http://pubs.acs.org/doi/abs/10.1021/ja964352a - ^ S. P. Cramer, F. M. F. deGroot, Y. Ma, C. T. Chen, F. Sette, C. A. Kipke, D. M. Eichhorn, M. K. Chan, W. H. Armstrong, E. Libby, G. Christou, S. Brooker, V. Mckee, O. C. Mullins, J. C. Fuggle. "Ligand field strengths and oxidation states from manganese L-edge spectroscopy" J. Am. Chem. Soc. 1991, 113, pp. 7937-7940.
http://pubs.acs.org/doi/abs/10.1021/ja00021a018 - ^ a b E. C. Wasinger, F. M. F. de Groot, B. Hedman, K. O. Hodgson, E. I. Solomon. "L-edge X-ray Absorption Spectroscopy of Non-Heme Iron Sites: Experimental Determination of Differential Orbital Covalency" J. Am. Chem. Soc. 2003, 125, pp. 12894–12906.
http://pubs.acs.org/doi/abs/10.1021/ja034634s - ^ a b R. K. Hocking, E. C. Wasinger, F. M. F. de Groot, K. O. Hodgson, B. Hedman, E. I. Solomon. "Fe L-Edge XAS Studies of K4[Fe(CN)6] and K3[Fe(CN)6]: A Direct Probe of Back-Bonding" J. Am. Chem. Soc. 2006, 128, pp. 10442-10451.
http://pubs.acs.org/doi/abs/10.1021/ja061802i
Categories:- Synchrotron-related techniques
- X-rays
- ^ T. E. Westre, P. Kennepohl, J. G. DeWitt, B. Hedman, K. O. Hodgson, E. I. Solomon. "A Multiplet Analysis of Fe K-Edge 1s → 3d Pre-Edge Features of Iron Complexes" J. Am. Chem. Soc. 1997, 119, pp. 6297-6314.


Wikimedia Foundation. 2010.