- Ene reaction
-
The Ene reaction (also known as the Alder-ene reaction) is a chemical reaction between an alkene with an allylic hydrogen (the ene) and a compound containing a multiple bond (the enophile), in order to form a new σ-bond with migration of the ene double bond and 1,5 hydrogen shift. The product is a substituted alkene with the double bond shifted to the allylic position. [1]
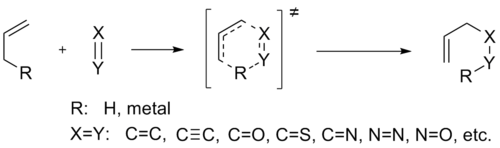
The ene reaction is mechanistically related to the Diels-Alder reaction, but the activation energies are much higher. As a result, ene reactions usually require high temperatures, which has limited their mechanistic exploration and synthetic use. [2] Nonetheless, many useful Lewis-Acid-catalyzed ene reactions have been developed that can afford high yields and selectivities at significantly lower temperatures. In general, the ene process is favored by electron-withdrawing substituents in the enophile, by strain in the ene component, and when the two reactants are aligned in a favorable geometry. [3]
Contents
The Ene Component
Enes are π-bonded molecules that contain at least one active hydrogen atom at the allylic, propargylic, or α-position. Possible ene components include olefinic, acetylenic, allenic, aromatic, cyclopropyl, and carbon-hetero bonds. [4] Usually, the allylic hydrogen of allenic components participates in ene reactions, but in the case of allenyl silanes, the allenic hydrogen atom α to the silicon substituent is the one transferred, affording a silylalkyne. Phenol can act as an ene component, for example in the reaction with dihydropyran, but high temperatures are required (150-170oC). Nonetheless, strained enes and fused small ring systems undergo ene reactions at much lower temperatures. In addition, ene components containing C=O, C=N and C=S bonds have been reported, but such cases are rare.[4]
The Enophile
Enophiles are π-bonded molecules which have electron-withdrawing substituents that lower significantly the LUMO of the π-bond. Possible enophiles contain carbon-carbon multiple bonds (olefins, acetylenes, benzynes), carbon-hetero multiple bonds (C=O in the case of carbonyl-ene reactions, C=N, C=S, C≡P), hetero-hetero multiple bonds (N=N, O=O, Si=Si, N=O, S=O), cumulene systems (N=S=O, N=S=N, C=C=O, C=C=S, SO2) and charged π systems (C=N+, C=S+, C≡O+, C≡N+).[4]
Mechanism
Concerted Pathway and Transition States
The mode of addition of the enophile to the ene in thermal ene reactions may be described as a suprafacial, three-component interaction among the HOMO of the ene, the LUMO of the allylic C-H bond of the ene, and the LUMO of the enophile (Figure 2). [5]
 Figure 2. Concerted Mechanism for the Ene Reaction.
Figure 2. Concerted Mechanism for the Ene Reaction.
The concerted nature of the ene process has been supported experimentally, [6] and the reaction can be designated as [σ2s + π2s + π2s] in the Woodward-Hoffmann notation. [5] The early transition state proposed for the thermal ene reaction of propene with formaldehyde has an envelope conformation, with a C-O-H angle of 155o, as calculated at the 3-21G level of theory.[7] Figure 3.View of the 3-21G Transition State Structure of the Propene- Formaldehyde Ene Reaction.
Figure 3.View of the 3-21G Transition State Structure of the Propene- Formaldehyde Ene Reaction.
The study of Lewis acid promoted carbonyl ene reactions, such as aluminum-catalyzed glyoxylate-ene processes (Figure 4), prompted researchers to consider a chair-like conformation for the transition state of ene reactions which proceed with relatively late transition states. [3] The advantage of such a model is the fact that steric parameters such as 1,3-diaxial and 1,2-diequatorial repulsions are easy to visualize, which allows for accurate predictions regarding the diastereoselectivity of many reactions. [3] Figure 4. Chair-like Transition State Proposed for Lewis-Acid Catalized Carbonyl-Ene Additions.
Figure 4. Chair-like Transition State Proposed for Lewis-Acid Catalized Carbonyl-Ene Additions.Biradical Mechanism
When a concerted mechanism is geometrically unfavorable, a thermal ene reaction can occur through a stepwise biradical pathway. For example, the ene reaction of cyclopentene and cyclohexene with diethyl azodicarboxylate can be catalyzed by free-radical initiators. The stepwise nature of the process is favored by the stability of the cyclopentenyl and cyclohexenyl radicals, as well as the difficulty of cyclopentene and cyclohexene in achieving the optimum geometry for a concerted process.[8][clarification needed]
 Figure 5. Stepwise, Biradical Pathway for the Ene Reaction.
Figure 5. Stepwise, Biradical Pathway for the Ene Reaction.Regioselection
Just as in the case of any cycloaddition, the success of an ene reaction is largely determined by the steric accessibility of the ene allylic hydrogen. In general, methyl and methylene H atoms are abstracted much more easily than methine hydrogens. In thermal ene reactions, the order of reactivity for the abstracted H atom is primary> secondary> tertiary, irrespective of the thermodynamic stability of the internal olefin product. In Lewis-acid promoted reactions, the pair enophile/Lewis acid employed determines largely the relative ease of abstraction of methyl vs. methylene hydrogens. [3]
The orientation of ene addition can be predicted from the relative stabilization of the developing partial charges in an unsymmetrical transition state with early formation of the σ bond. The major regioisomer will come from the transition state in which transient charges are best stabilized by the orientation of the ene and enophile. [4]
Internal Asymmetric Induction
In terms of the diastereoselection with respect to the newly created chiral centers, an endo preference has been qualitatively observed, but steric effects can easily modify this preference (Figure 6). [3]
 Fugure 6. Endo Preference for the Ene Reaction.
Fugure 6. Endo Preference for the Ene Reaction.Intramolecular Ene Reactions
Intramolecular ene reactions benefit from less negative entropies of activation than their intrermolecular counterparts, so are usually more facile, occurring even in the case of simple enophiles, such as unactivated alkenes and alkynes. [9]. The high regio- and stereoselectivities that can be obtained in these reactions can offer considerable control in the synthesis of intricate ring systems.
Considering the position of attachment of the tether connecting the ene and enophile, Oppolzer [3] has classified both thermal and Lewis acid-catalyzed intramolecular ene reactions as types I, II and III, and Snider [2] has added a type IV reaction (Figure 7). In these reactions, the orbital overlap between the ene and enophile is largely controlled by the geometry of the approach of components [4].
 Figure 7. Types of Intramolecular Ene Reactions.
Figure 7. Types of Intramolecular Ene Reactions.Lewis Acid - Catalyzed Ene Reactions
Advantages and Rationale
Thermal ene reactions have several drawbacks, such as the need for very high temperatures and the possibility of side reactions, like proton-catalyzed olefin polymerization or isomerization reactions. Since enophiles are electron-deficient, it was reasoned that their complexation with Lewis acids should accelerate the ene reaction, as it occurred for the reaction shown in Figure 8.
 Figure 8. Improvements Brought to the Ene Reaction by Lewis Acid Catalysis.
Figure 8. Improvements Brought to the Ene Reaction by Lewis Acid Catalysis.Alykaluminum halides are well known as proton scavengers, and their use as Lewis acid catalysts in ene reactions has greatly expanded the scope of these reactions and has allowed their study and development under significantly milder conditions.[2]
Since a Lewis acid can directly complex to a carbonyl oxygen, numerous trialkylaluminum catalysts have been developed for enophiles that contain a C=O bond. In particular, it was found that Me2AlCl is a very useful catalyst for the ene reactions of α,β-unsaturated aldehydes and ketones, and other aliphatic and aromatic aldehydes. The reason behind the success of this catalyst is the fact that the ene-adduct- Me2AlCl complex can further react to afford methane and aluminum alkoxide, which can prevent proton-catalyzed rearrangements and solvolysis (Figure 9).[2]
 Figure 9. Me2AlCl-Catalyzed Carbonyl-Ene Reactions.
Figure 9. Me2AlCl-Catalyzed Carbonyl-Ene Reactions.
In the case of directed carbonyl-ene reactions, high levels of regio- and stereoselectivity have been observed upon addition of a Lewis Acid, which can be explained through chair-like transition states. Notably, some of these reactions (Figure 10) can run at very low temperatures and still afford very good yields of a single regioisomer [3]. Figure 10. Lewis Acid-Catalyzed Directed Carbonyl-Ene Reaction.
Figure 10. Lewis Acid-Catalyzed Directed Carbonyl-Ene Reaction.Reaction Conditions
As long as the nucleophilicity of the alkyl group does not lead to side reactions, catalytic amounts of Lewis acid are sufficient for many ene reactions with reactive enophiles. Nonetheless, the amont of Lewis acid can widely vary, as it largely depends on the relative basicity of the enophile and the ene adduct. In terms of solvent choice for the reactions, the highest rates are usually achieved using halocarbons as solvents; polar solvents such as ethers are not suitable, as they would complex to the Lewis acid, rendering the catalyst inactive. [2]
Reactivity of Enes
While steric effects are still important in determining the outcome of a Lewis acid catalyzed ene reaction, electronic effects are also significant, since in such a reaction, there will be a considerable positive charge developed at the central carbon of the ene. As a result, alkenes with at least one disubstituted vinylic carbon are much more reactive than mono or 1,2 disubstituted ones.
Mechanism
As seen in Figure 11, Lewis acid catalyzed ene reactions can proceed either through a concerted mechanism that has a polar transition state, or through a stepwise mechanism with a zwitterionic intermediate. The ene, enophile and choice of catalyst can all influence which pathway is the lower energy process. In general, the more reactive the ene or enophile-Lewis acid complex is, the more likely the reaction is to be stepwise. [2]
 Figure 11. Mechanisms of Lewis Acid Catalyzed Ene Reactions.
Figure 11. Mechanisms of Lewis Acid Catalyzed Ene Reactions.Asymmetric catalysis
A current direction in the study of Lewis acid-catalyzed ene reactions is the developemnt of asymmetric catalysts for C-C bond formation. One of the first examples of such a catalyst is the chiral titanium complex (R)-1a, which was developed by Mikami [10] for asymmetric ene reactions involving prochiral glyoxylate. The catalyst is prepared in situ from (i-PrO)2TiX2 and optically pure binaphthol, the alkoxy-ligand exchange being facilitated by the use of molecular sieves. The method affords α-hydroxy esters of high enantiomeric purities, compounds that represent a class of biological and synthetic importance (Figure 12). [10]
 Figure 12. Asymmetric Glyoxylate-Ene Reaction Catalyzed by a Chiral Titanium Complex.
Figure 12. Asymmetric Glyoxylate-Ene Reaction Catalyzed by a Chiral Titanium Complex.Since both (R)- and (S)-BINOL are commercially available in optically pure form, this asymmetric process allows the synthesis of both enantiomers of α-hydroxy esters and their derivatives. However, this method is not applicable to mono and 1,2-disubstituted olefins, since such compounds cannot undergo ene reaction with glyoxylate. [10]
The formal total synthesis of laulimalide [11] (compound 1, Figure 13) illustrates how powerful and robust the reaction developed by Mikami is. Laulimalide is a marine natural product, a metabolite of various sponges that could find a potential use as an anti-tumor agent, due to its ability to stabilize microtubuli. One of the key steps in the strategy used for the synthesis of the C3-C16 fragment was a chirally catalyzed ene reaction that installed the C15 stereocenter. Treatment of the terminal allyl group of compound 3 with ethyl glyoxylate in the presence of catalytic (S)-BINOL-TiBr2 provided the required alcohol in 74% yield and >95% ds. This method eliminated the need for a protecting group or any other functionality at the end of the molecule. In addition, by carrying out this reaction, Pitts et al. managed to avoid the harsh conditions and low yields associated with installing exo-methylene units late in the synthesis. [11]
 Figure 13. Retrosynthetic Analysis of the C3-C16 Fragment of Laulimalide.
Figure 13. Retrosynthetic Analysis of the C3-C16 Fragment of Laulimalide.See also
- Diels-Alder reaction
- Certain isotoluenes isomerize by an ene mechanism
References
- ^ Alder, K.; Pascher, F; Schmitz, A. (1943). Ber. dtsch. chem. Ges 76: 27.
- ^ a b c d e f Snider, B. B. (1980). Acc. Chem. Res. 13: 426.
- ^ a b c d e f g Mikami, K.; Shimizu, M. (1992). Chem. Rev. 92: 1021.
- ^ a b c d e Paderes, G. D.; Jorgensen, W. L. (1992). J. Org. Chem. 57: 1904 and references within.
- ^ a b Inagaki, S.; Fujimoto, H; Fukui, K. J. (1976). J. Am. Chem. Soc. 41: 4693.
- ^ Stephenson, L. M.; Mattern, D. L. (1976). J. Org. Chem. 41: 3614.
- ^ Loncharich, R. J.; Houk, K. N. (1987). J. Am. Chem. Soc. 109: 6947.
- ^ Thaler, W. A.; Franzus, B. J. (1964). J. Org. Chem. 29: 2226.
- ^ Oppolzer, W.; Snieckus, V. (1978). Angew. Chem. Int. Ed. Engl. 17: 476.
- ^ a b c Mikami, K.; Terada, M.; Takeshi, N. (1990). J. Am. Chem. Soc. 112: 3949.
- ^ a b Pitts, M. R.; Mulzer, J. (2002). Tetrahedron Letters 43: 8471.
Categories:- Carbon-carbon bond forming reactions
- Rearrangement reactions
- Reaction mechanisms

Wikimedia Foundation. 2010.