- Crich beta-mannosylation
-
The Crich β-mannosylation is a synthetic strategy which is used in carbohydrate synthesis to generate a 1,2-cis-glycosidic bond. This type of linkate is generally very difficult to make, and specific methods like the Crich β-mannosylation are used to overcome these issues.
Contents
Background
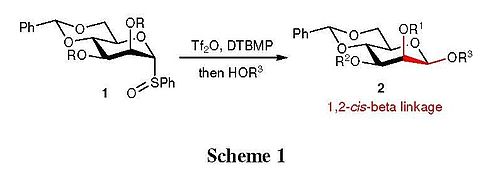
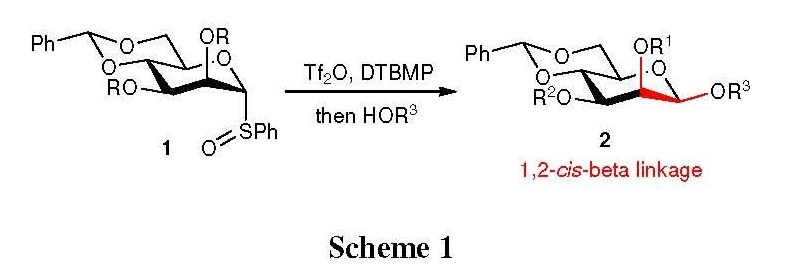
The development of facile chemical glycosylation protocols is essential important in synthesizing complex oligosaccharides. Among many diverse type of glycosidic linkages, the 1,2-cis-β-glycoside, which exists in many biologically relevant glycoconjugates and oligosaccharides, is arguably one of the most difficult to synthesize.[1] The challenges in constructing β-mannose linkage have been well documented in several reviews.[2][3] To date, a few laboratories have devised efficient methodologies to overcome these synthetic hurdles, and achieved varying degrees of success. Of those elegant approaches, a highly stereoselective β-mannosylation protocol developed by Crich and co-workers was realized as a breakthrough in β-mannoside synthesis.[4][5][6] This strategy is based on the initial activation of α-mannosyl sulfoxides 1 with triflic anhydride (Tf2O) using DTBMP (2,6-di-tert-butyl-4-methylpyridine) as a base, followed by nucleophilic substitution of glycosyl acceptors (HOR3) to provide the 1,2-cis-β-glycoside 2 in good yield and selectivity (Scheme 1).
Mechanistic Studies
The mechanistic details of this reaction have been extensively explored by Crich’s laboratories.[7][8] Low-temperature 1H, 13C, and 19F NMR spectroscopic investigations revealed that anomeric triflate 3 derived from 1 is the intermediate glycosyl donor. Moreover, the mechanism of glycosidic bond forming reaction (3→2) was examined thoroughly by the determination of kinetic isotopic effects (KIEs) and NMR spectroscopy. Consequently, the magnitude of KIEs indicated that the displacement of the triflate from 3 proceeded with the development of significant oxacarbenium ion character at the anomeric position. This might be rationalized either by (1) a dissociative mechanism involving the intermediacy of either a transient contact ion pair (CIP) 4 or a solvent-separated ion pair (SSIP) 5, or (2) a mechanistically variant transition state 7 (Scheme 2).
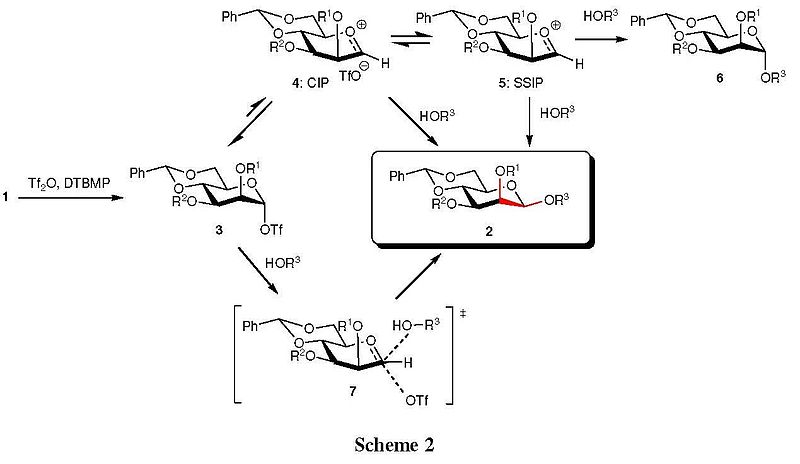
For the intermediate CIP 4, the triflate anion is closely associated with face where it just departed thus shields that side against nucleophilic attack. For the alternative intermediate SSIP 5 which is in equilibrium with an initial CIP, the anomeric center could presumably be attacked by incoming alcohol from either face, giving β-mannoside 2 along with the undesired α-anomer 6. Along these lines, the presence of the 4,6-O-benzylidene protecting group, which serves to rigidify the pyranoside against rehybridization at the anomeric carbon, is essential in shifting the equilibrium toward the covalent triflate, thus reducing α-glycoside formation. Additionally, the only intermediate observed by NMR spectroscopy is the covalent triflate 3, indicating that the complete set of equilibria between 3, the CIP 4, and SSIP 5 set is very heavily biased towards 3.
Reaction Scope
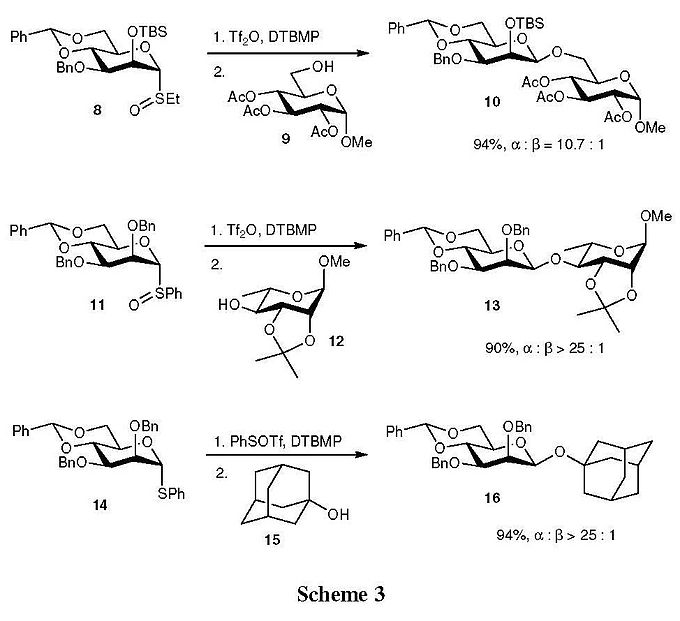
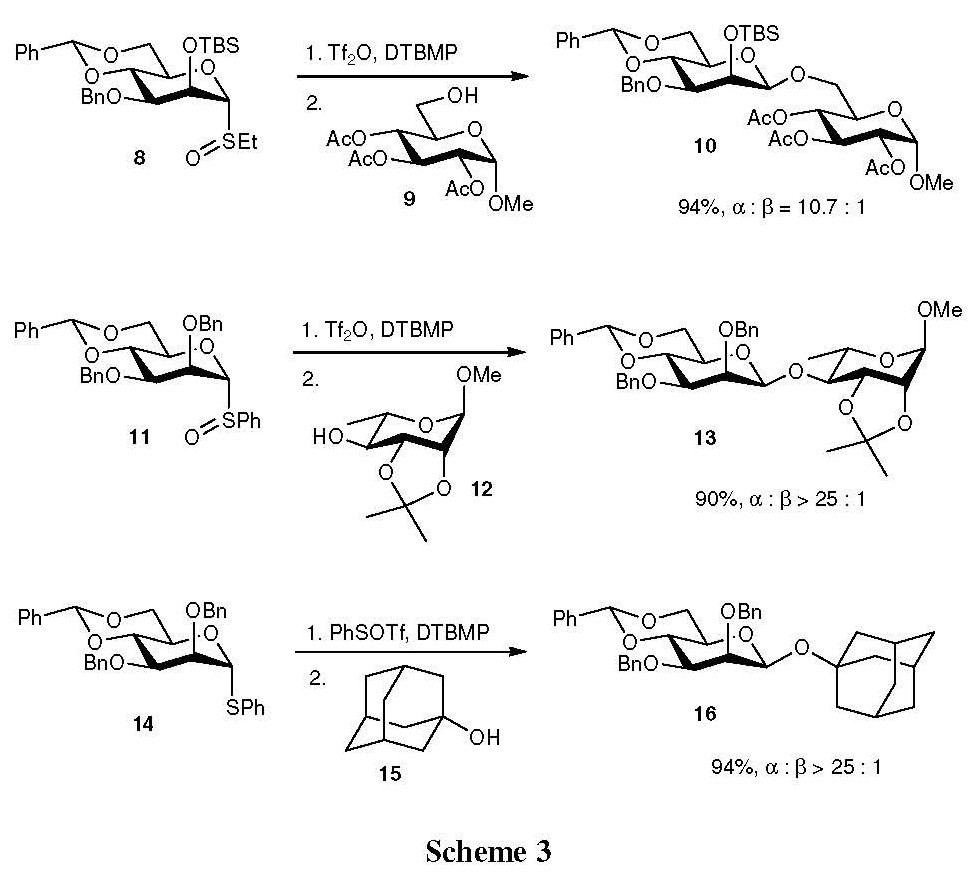
Some representative examples of Crich’s β-mannosylation are shown in Scheme 3.[9] It is noteworthy that, with this method in hand, primary, secondary, and tertiary alcohols (9, 12, and 13) all serve as glycosyl acceptors effectively in terms of yields and selectivity. In a recent version, the β-mannosylation of thioglycoside 14 and its analogues were examined to prepare sterically hindered glycosides, in which PhSOTf (or other newly developed sulfur-type oxidants[10][11]) served as a convenient reagent for the in situ generation of the glycosyl triflate from 14, thus facilitating the reaction.
Solid-Phase Synthesis
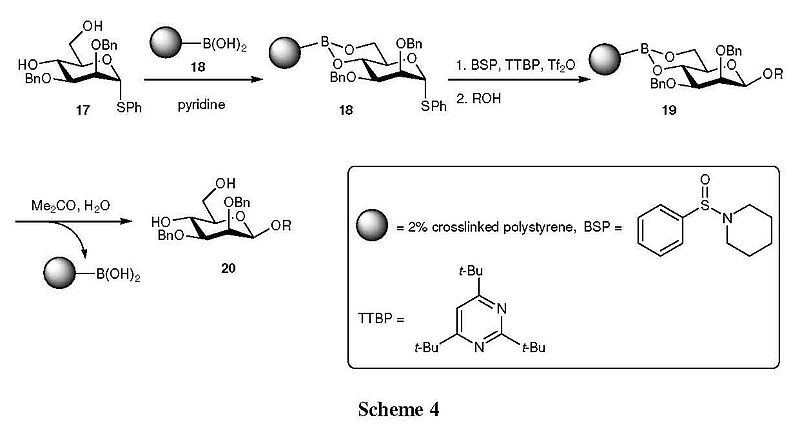
The polymer-supported synthesis of β-mannosides based on the Crich’s protocol has also been studied in the same laboratories.[12] As shown in Scheme 4, diol 17 was first reacted with polystyrylboronic acid (18) to offer the bound donor 19, in which 4,6-O-phenylboronates served as the torsionally disarming protecting group. With that, activation of the thioglycoside 19 was readily achieved, and the coupling reaction with the acceptor alcohol underwent smoothly to provide the bound β-mannoside 20. After removal of the excess reagents and byproducts from the resin, 20 was then treated with aqueous acetone to release 4,6-diol 21. Overall, this is a powerful method for solid-phase synthesis of β-mannosides, which has great potential to be further extended, was established.
See also
- Carbohydrate synthesis
- Difficult linkages
- Carbohydrate chemistry
References
- ^ Gridley, J. J.; Osborn, H. M. I. J. Chem. Soc., Perkin Trans. 1 2000, 1471.
- ^ Kaji, E.; Lichtenthaler, F. W. Trends Glycosci. Glycotechnol. 1993, 5, 121.
- ^ Banoub, J. Chem. Rev. 1992, 92, 1167.
- ^ Crich, D.; Sun, S. J. Org. Chem. 1996, 61, 4506.
- ^ Crich, D.; Sun, S. J. Org. Chem. 1997, 62, 1198.
- ^ Crich, D.; Sun, S. J. Am. Chem. Soc. 1998, 120, 435.
- ^ Crich, D.; Sun, S. J. Am. Chem. Soc. 1997, 119, 11217.
- ^ Crich, D.; Chandrasekera, N. S. Angew. Chem. Int. Ed. 2004, 43, 5386.
- ^ Crich, D.; Sun, S. Tetrahedron 1998, 54, 8321.
- ^ Crich, D.; Smith, M. Org. Lett. 2000, 2, 4067.
- ^ Crich, D.; Smith, M. J. Am. Chem. Soc. 2001, 123, 9015.
- ^ Crich, D.; Smith, M. J. Am. Chem. Soc. 2002, 124, 8867.
Categories:- Carbohydrate chemistry
- Organic reactions


Wikimedia Foundation. 2010.