- MECP2
-
MECP2 (methyl CpG binding protein 2 (Rett syndrome)) is a gene[1] that provides instructions for making its protein product, MECP2, also referred to as MeCP2.[2] MECP2 appears to be essential for the normal function of nerve cells. The protein seems to be particularly important for mature nerve cells, where it is present in high levels. The MeCP2 protein is likely to be involved in turning off ("repressing" or "silencing") several other genes. This prevents the genes from making proteins when they are not needed. Recent work has shown that MeCP2 can also activate other genes.[3] The MECP2 gene is located on the long (q) arm of the X chromosome in band 28 ("Xq28"), from base pair 152,808,110 to base pair 152,878,611.
DNA methylation is the major modification of eukaryotic genomes and plays an essential role in mammalian development. Human proteins MECP2 (this protein), MBD1, MBD2, MBD3, and MBD4 comprise a family of nuclear proteins related by the presence in each of a methyl-CpG binding domain (MBD). Each of these proteins, with the exception of MBD3, is capable of binding specifically to methylated DNA. MECP2, MBD1 and MBD2 can also repress transcription from methylated gene promoters. In contrast to other MBD family members, MECP2 is X-linked and subject to X inactivation. MECP2 is dispensible in stem cells. MECP2 gene mutations are the cause of most cases of Rett syndrome, a progressive neurologic developmental disorder and one of the most common causes of mental retardation in females.[4]
Contents
Function
MeCP2 protein is found in all cells in the body, including the brain, acting as a transcriptional repressor and activator, depending on the context. However, the idea that MeCP2 functions as an activator is relatively new and remains controversial.[5] In the brain, it is found in high concentrations in neurons and is associated with maturation of the central nervous system (CNS) and in forming synaptic contacts.[6]
Mechanism of action
The MeCP2 protein binds to forms of DNA that have been methylated. The MeCP2 protein then interacts with other proteins to form a complex that turns off the gene. Methylation is a chemical alteration made to a "cytosine" (C) when it occurs in a particular DNA sequence, "CpG". Many genes have CpG islands, which frequently occur near the beginning of the gene. MECP2 does not bind to these islands in most cases, as they are not methylated. The expression of a few genes may be regulated through methylation of their CpG island, and MECP2 may play a role in a subset of these. Researchers have not yet determined which genes are targeted by the MeCP2 protein, but such genes are probably important for the normal function of the central nervous system. However, the first large-scale mapping of MECP2 binding sites in neurons found that only 6% of the binding sites are in CpG islands, and that 63% of MECP2-bound promoters are actively expressed and only 6% are highly methylated, indicating that MECP2's main function is something other than silencing methylated promoters.[7]
Once bound, MeCP2 will condense the chromatin structure, form a complex with histone deacetylases (HDAC), or block transcription factors directly. More recent studies have demonstrated that MeCP2 may also function as a transcriptional activator, through recruiting the transcription factor CREB1. This was an unexpected finding which suggests that MeCP2 is a key transcriptional regulator with potentially dual roles in gene expression. In fact, the majority of genes that are regulated by MeCP2 appear to be activated rather than repressed.[8] However, it remains controversial whether MeCP2 regulates these genes directly or whether these changes are secondary in nature.[5] Further studies have shown MeCP2 may be able to bind directly to un-methylated DNA in some instances.[9] MeCP2 has been implicated in regulation of imprinted genes and loci that include UBE3A and DLX5.[10]
Structure
MeCP2 is part of a family of methyl-CpG-binding domain proteins (MBD), but possesses its own unique differences which help set it apart from the group. It has two functional domains:
- a methyl-cytosine-binding domain (MBD) composed of 85 amino acids; and
- a transcriptional repression domain (TRD) composed of 104 amino acids
The MBD domain forms a wedge and attaches to the methylated CpG sites on the DNA strands. The TRD region then reacts with SIN3A to recruit histone deacetylases (HDAC).[11] There are also unusual, repetitive sequences found at the carboxyl terminus. This region is closely related to the fork head family, at the amino acid level.[12]
Role in disease
Rett syndrome is caused by mutations in the MECP2 gene. Several types of mutations have been identified in people with Rett syndrome. These mutations include changes in single base pairs (the building material of DNA), insertions or deletions of DNA in the gene, and changes that affect how the gene is processed into a protein. Mutations in the gene alter the structure of the MeCP2 protein or lead to reduced amounts of the protein. As a result, the protein is unable to bind to DNA or turn other genes on or off. Genes that are normally repressed by MeCP2 remain active and continue to make large amounts of particular proteins when they are not needed. Other genes that are normally activated by MeCP2 remain inactive and thus unable to make protein products. This defect probably disrupts the normal functioning of nerve cells, leading to the signs and symptoms of Rett syndrome.
This disease is mainly found in girls with a prevalence of around 1 in every 10,000. Patients are born with very hard to find signs of a disorder, but after about six months to a year and half, speech and motor function capabilities start to decrease. This is followed by seizures, growth retardation and cognitive and motor impairment.[13] This is a X-linked dominant disease that is found predominantly affecting the paternal X chromosome. It has been linked to male lethality, due to its prevalence in females, but in rare cases some males can also be affected by Rett Syndrome.[14]
Mutations in the MECP2 gene have also been identified in people with several other disorders affecting the central nervous system. For example, MECP2 mutations are associated with some cases of moderate to severe X-linked mental retardation. Mutations in the gene have also been found in males with severe brain dysfunction (neonatal encephalopathy) who live only into early childhood. In addition, several people with features of both Rett syndrome and Angelman syndrome (a condition characterized by mental retardation, problems with movement, and inappropriate laughter and excitability) have mutations in the MECP2 gene. Lastly, MECP2 mutations or changes in the gene's activity have been reported in some cases of autism (a developmental disorder that affects communication and social interaction).
More recent studies reported genetic polymorphisms in the MeCP2 genes in patients with systemic lupus erythematosus (SLE).[15] SLE is a systemic autoimmune disease that can affect multiple organs. MeCP2 polymorphisms have been reported so far in European-derived and Asian lupus patients.
The genetic loss of MECP2 has been identified as changing the properties of cells in the locus ceruleus the exclusive source of noradrenergic innervation to the cerebral cortex and hippocampus.[16]
Researchers have concluded that "Because these neurons are a pivotal source of norepinephrine throughout the brainstem and forebrain and are involved in the regulation of diverse functions disrupted in Rett syndrome, such as respiration and cognition, we hypothesize that the locus ceruleus is a critical site at which loss of MECP2 results in CNS dysfunction."[16]
Interactions
MECP2 has been shown to interact with SKI protein[17] and Nuclear receptor co-repressor 1.[17] In neuronal cells the MECP2 mRNA is thought to interact with miR-132, which silences the expression of the protein. This forms part of a homeostatic mechanism that could regulate MECP2 levels in the brain.[18]
References
- ^ Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (October 1999). "Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2". Nat. Genet. 23 (2): 185–8. doi:10.1038/13810. PMID 10508514.
- ^ Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A (June 1992). "Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA". Cell 69 (6): 905–14. doi:10.1016/0092-8674(92)90610-O. PMID 1606614.
- ^ Chahrour M, et al. (2008). "MeCP2, a key contributor to neurological disease, activates and represses transcription". Science 320 (5880): 1224–9. doi:10.1126/science.1153252. PMC 2443785. PMID 18511691. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2443785.
- ^ "Entrez Gene: MECP2 methyl CpG binding protein 2 (Rett syndrome)". http://www.ncbi.nlm.nih.gov/sites/entrez?Db=gene&Cmd=ShowDetailView&TermToSearch=4204.
- ^ a b Cohen S, Zhou Z, Greenberg ME (May 2008). "Medicine. Activating a repressor.". Science 320 (5880): 1172–3. doi:10.1126/science.1159146. PMC 2857976. PMID 18511680. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2857976.
- ^ Luikenhuis S, Giacometti E, Beard CF, Jaenisch R (April 2004). "Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice". Proc. Natl. Acad. Sci. U.S.A. 101 (16): 6033–8. doi:10.1073/pnas.0401626101. PMC 395918. PMID 15069197. http://www.pnas.org/cgi/content/full/101/16/6033.
- ^ Yasui DH, Peddada S, Bieda MC, Vallero RO, Hogart A, Nagarajan RP, Thatcher KN, Farnham PJ, Lasalle JM (December 2007). "Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes". Proc. Natl. Acad. Sci. U.S.A. 104 (49): 19416–21. doi:10.1073/pnas.0707442104. PMC 2148304. PMID 18042715. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2148304.
- ^ Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY (May 2008). "MeCP2, a key contributor to neurological disease, activates and represses transcription". Science 320 (5880): 1224–9. doi:10.1126/science.1153252. PMC 2443785. PMID 18511691. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2443785.
- ^ Georgel PT, Horowitz-Scherer RA, Adkins N, Woodcock CL, Wade PA, Hansen JC (August 2003). "Chromatin compaction by human MeCP2. Assembly of novel secondary chromatin structures in the absence of DNA methylation". J. Biol. Chem. 278 (34): 32181–8. doi:10.1074/jbc.M305308200. PMID 12788925.
- ^ LaSalle JM (2007). "The odyssey of MeCP2 and parental imprinting". Epigenetics 2 (1): 5–10. doi:10.4161/epi.2.1.3697. PMC 1866173. PMID 17486180. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1866173.
- ^ Wakefield RI, Smith BO, Nan X, Free A, Soteriou A, Uhrin D, Bird AP, Barlow PN (September 1999). "The solution structure of the domain from MeCP2 that binds to methylated DNA". J. Mol. Biol. 291 (5): 1055–65. doi:10.1006/jmbi.1999.3023. PMID 10518942.
- ^ Paul A. Wade
- ^ Caballero IM, Hendrich B (April 2005). "MeCP2 in neurons: closing in on the causes of Rett syndrome". Hum. Mol. Genet. 14 Spec No 1: R19–26. doi:10.1093/hmg/ddi102. PMID 15809268.
- ^ Samaco RC, Nagarajan RP, Braunschweig D, LaSalle JM (March 2004). "Multiple pathways regulate MeCP2 expression in normal brain development and exhibit defects in autism-spectrum disorders". Hum. Mol. Genet. 13 (6): 629–39. doi:10.1093/hmg/ddh063. PMID 14734626.
- ^ Sawalha AH, Webb R, Han S, Kelly JA, Kaufman KM, Kimberly RP, Alarc?n-Riquelme ME, James JA, Vyse TJ, Gilkeson GS, Choi CB, Scofield RH, Bae SC, Nath SK, Harley JB (2008). Jin, Dong-Yan. ed. "Common variants within MECP2 confer risk of systemic lupus erythematosus". PLoS ONE 3 (3): e1727. doi:10.1371/journal.pone.0001727. PMC 2253825. PMID 18320046. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2253825.
- ^ a b Taneja P, Ogier M, Brooks-Harris G, Schmid DA, Katz DM, Nelson SB (2009). "Pathophysiology of Locus Ceruleus Neurons in a Mouse Model of Rett Syndrome". Journal of Neuroscience 29 (39): 12187–12195. doi:10.1523/JNEUROSCI.3156-09.2009. PMC 2846656. PMID 19793977. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2846656.
- ^ a b Kokura, K; Kaul S C, Wadhwa R, Nomura T, Khan M M, Shinagawa T, Yasukawa T, Colmenares C, Ishii S (Sep. 2001). "The Ski protein family is required for MeCP2-mediated transcriptional repression". J. Biol. Chem. (United States) 276 (36): 34115–21. doi:10.1074/jbc.M105747200. ISSN 0021-9258. PMID 11441023.
- ^ Klein ME, Lioy DT, Ma L, Impey S, Mandel G, Goodman RH (2007). "Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA". Nat Neurosci 10 (12): 1513–4. doi:10.1038/nn2010. PMID 17994015.
Further reading
- Chahrour M, Zoghbi HY (2007). "The story of Rett syndrome: from clinic to neurobiology". Neuron 56 (3): 422–37. doi:10.1016/j.neuron.2007.10.001. PMID 17988628.
- Carney RM, Wolpert CM, Ravan SA, Shahbazian M, Ashley-Koch A, Cuccaro ML, Vance JM, Pericak-Vance MA (2003). "Identification of MeCP2 mutations in a series of females with autistic disorder". Pediatr Neurol 28 (3): 205–11. PMID 12770674.
- Kerr AM, Ravine D (2003). "Review article: breaking new ground with Rett syndrome". J Intellect Disabil Res 47 (Pt 8): 580–7. doi:10.1046/j.1365-2788.2003.00506.x. PMID 14641805.
- Neul JL, Zoghbi HY (2004). "Rett syndrome: a prototypical neurodevelopmental disorder". Neuroscientist 10 (2): 118–28. doi:10.1177/1073858403260995. PMID 15070486.
- Schanen C, Houwink EJ, Dorrani N, Lane J, Everett R, Feng A, Cantor RM, Percy A (2004). "Phenotypic manifestations of MECP2 mutations in classical and atypical Rett syndrome". Am J Med Genet A 126 (2): 129–40. doi:10.1002/ajmg.a.20571. PMID 15057977.
- Van den Veyver IB, Zoghbi HY (2001). "Mutations in the gene encoding methyl-CpG-binding protein 2 cause Rett syndrome". Brain Dev 23 (Suppl 1): S147–51. doi:10.1016/S0387-7604(01)00376-X. PMID 11738862.
- Webb T, Latif F (2001). "Rett syndrome and the MECP2 gene". J Med Genet 38 (4): 217–23. doi:10.1136/jmg.38.4.217. PMC 1734858. PMID 11283201. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1734858.
- Shahbazian MD, Zoghbi HY (2003). "Rett syndrome and MeCP2: linking epigenetics and neuronal function.". Am. J. Hum. Genet. 71 (6): 1259–72. doi:10.1086/345360. PMC 378559. PMID 12442230. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=378559.
- Moog U, Smeets EE, van Roozendaal KE, et al. (2003). "Neurodevelopmental disorders in males related to the gene causing Rett syndrome in females (MECP2).". Eur. J. Paediatr. Neurol. 7 (1): 5–12. doi:10.1016/S1090-3798(02)00134-4. PMID 12615169.
- Miltenberger-Miltenyi G, Laccone F (2004). "Mutations and polymorphisms in the human methyl CpG-binding protein MECP2.". Hum. Mutat. 22 (2): 107–15. doi:10.1002/humu.10243. PMID 12872250.
- Weaving LS, Ellaway CJ, Gécz J, Christodoulou J (2006). "Rett syndrome: clinical review and genetic update.". J. Med. Genet. 42 (1): 1–7. doi:10.1136/jmg.2004.027730. PMC 1735910. PMID 15635068. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1735910.
- Bapat S, Galande S (2005). "Association by guilt: identification of DLX5 as a target for MeCP2 provides a molecular link between genomic imprinting and Rett syndrome.". Bioessays 27 (7): 676–80. doi:10.1002/bies.20266. PMID 15954098.
- Zlatanova J (2005). "MeCP2: the chromatin connection and beyond.". Biochem. Cell Biol. 83 (3): 251–62. doi:10.1139/o05-048. PMID 15959553.
- Kaufmann WE, Johnston MV, Blue ME (2006). "MeCP2 expression and function during brain development: implications for Rett syndrome's pathogenesis and clinical evolution.". Brain Dev. 27 (Suppl 1): S77–S87. doi:10.1016/j.braindev.2004.10.008. PMID 16182491.
- Armstrong DD (2006). "Can we relate MeCP2 deficiency to the structural and chemical abnormalities in the Rett brain?". Brain Dev. 27 (Suppl 1): S72–S76. doi:10.1016/j.braindev.2004.10.009. PMID 16182497.
- Santos M, Coelho PA, Maciel P (2006). "Chromatin remodeling and neuronal function: exciting links.". Genes, Brain and Behavior 5 (Suppl 2): 80–91. doi:10.1111/j.1601-183X.2006.00227.x. PMID 16681803.
- Bienvenu T, Chelly J (2006). "Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized.". Nat. Rev. Genet. 7 (6): 415–26. doi:10.1038/nrg1878. PMID 16708070.
- Francke U (2007). "Mechanisms of disease: neurogenetics of MeCP2 deficiency.". Nature clinical practice. Neurology 2 (4): 212–21. doi:10.1038/ncpneuro0148. PMID 16932552.
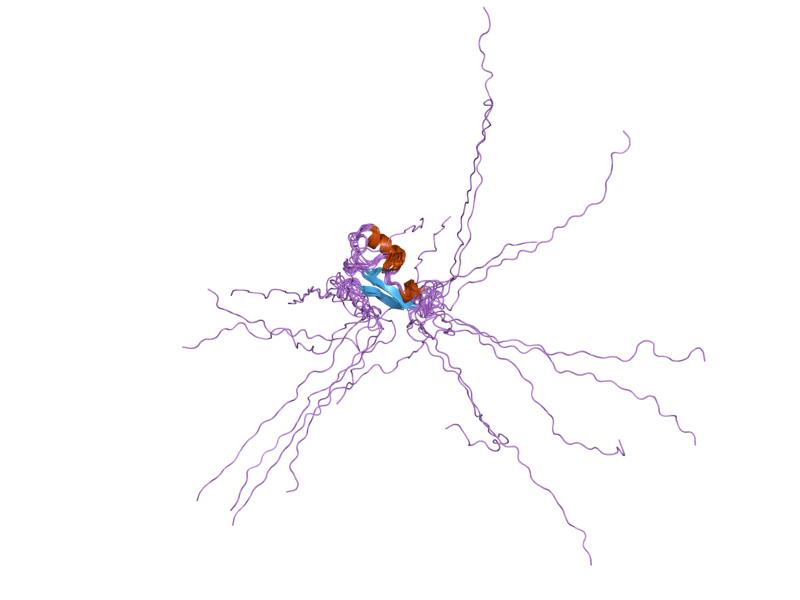
PDB gallery  1qk9: THE SOLUTION STRUCTURE OF THE DOMAIN FROM MECP2 THAT BINDS TO METHYLATED DNA
1qk9: THE SOLUTION STRUCTURE OF THE DOMAIN FROM MECP2 THAT BINDS TO METHYLATED DNA 1ub1: Solution structure of the matrix attachment region-binding domain of chicken MeCP2
1ub1: Solution structure of the matrix attachment region-binding domain of chicken MeCP2External links
Categories:- Human proteins
- Genes
- DNA-binding proteins
- Transcription factors


Wikimedia Foundation. 2010.