- NUPACK
-
NUPACK 
URL nupack.org Commercial? No Registration Optional Created by The NUPACK Team at Caltech The Nucleic Acid Package, is a growing software suite for the analysis and design of nucleic acid systems [1]. Jobs can be run online on the NUPACK webserver or NUPACK source code can be downloaded and compiled locally. NUPACK algorithms are formulated in terms of nucleic acid secondary structure. In most cases, pseudoknots are excluded from the structural ensemble.
Contents
Secondary structure model
 An example secondary structure drawing (left) and the corresponding polymer graph (right). Backbones are represented by thick colored lines and bases and base pairs are represented by thin black lines.
An example secondary structure drawing (left) and the corresponding polymer graph (right). Backbones are represented by thick colored lines and bases and base pairs are represented by thin black lines.The secondary structure of multiple interacting strands is defined by a list of base pairs [2]. A polymer graph for a secondary structure can be constructed by ordering the strands around a circle, drawing the backbones in succession from 5’ to 3’ around the circumference with a nick between each strand, and drawing straight lines connecting paired bases. A secondary structure is pseudoknotted if every strand ordering corresponds to a polymer graph with crossing lines. A secondary structure is connected if no subset of the strands is free of the others. Algorithms are formulated in terms of ordered complexes, each corresponding to the structural ensemble of all connected polymer graphs with no crossing lines for a particular ordering of a set of strands. The free energy of an unpseudoknotted secondary structure is calculated using nearest-neighbor empirical parameters for RNA in 1M Na+ [3][4] or for DNA in user-specified Na+ and Mg++ concentrations [5] [6] [7]; additional parameters are employed for the analysis of pseudoknots (single RNA strands only) [8] [9].
Web server
Analysis
The Analysis page allows users to analyze the thermodynamic properties of a dilute solution of interacting nucleic acid strands in the absence of pseudoknots (e.g., a test tube of DNA or RNA strand species) [1] [2]. For a dilute solution containing multiple strand species interacting to form multiple species of ordered complexes, NUPACK calculates for each ordered complex:
- the partition function,
- the minimum free energy (MFE) secondary structure,
- the equilibrium base-pairing probabilities,
- its equilibrium concentration,
including rigorous treatment of distinguishability issues that arise in the multi-stranded setting.
Design
The Design page allows users to design sequences for one or more strands intended to adopt an unpseudoknotted target secondary structure at equilibrium [1]. Sequence design is formulated as an optimization problem with the goal of reducing the ensemble defect below a user-specified stop condition [10]. For a candidate sequence and a given target secondary structure, the ensemble defect is the average number of incorrectly paired over the structural ensemble of the ordered complex [11]. For a target secondary structure with N nucleotides, the algorithm seeks to achieve an ensemble defect below N/100. Empirically, the design algorithm exhibits asymptotic optimality as N increases: for sufficiently large N, the cost of sequence design is typically only 4/3 the cost of a single evaluation of the ensemble defect [10].
Utilities
The Utilities page allows users to evaluate, display, and annotate the equilibrium properties of a complex of interacting nucleic acid strands [1]. The page accepts as input either sequence information, structure information, or both, performing diverse functions based on the information provided, including automatic layout and rendering of secondary structures with or without ideal helical geometry. In either case, the structure layout can be edited dynamically within the web application.
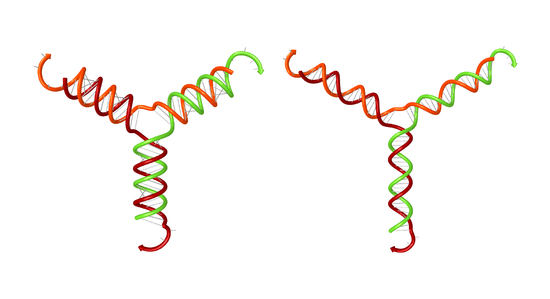 The Utilities page enables depiction of secondary structures with ideal helical geometry for stacked base pairs, as for this complex of three RNA strands with A-form helices (left) or three DNA strands with B-form helices (right).
The Utilities page enables depiction of secondary structures with ideal helical geometry for stacked base pairs, as for this complex of three RNA strands with A-form helices (left) or three DNA strands with B-form helices (right).
Implementation
The NUPACK web application [1] is programmed within the Ruby on Rails framework, employing AJAX and the Dojo Toolkit to implement dynamic features and interactive graphics. Plots and graphics are generated using NumPy and matplotlib. The site is supported on current versions of the Safari, Chrome, and Firefox browsers. The NUPACK library of analysis and design algorithms is written in the C programming language. Dynamic programs are parallelized using MPI.
Terms of use
The NUPACK web server and NUPACK source code are provided for non-commercial research purposes.
Funding
NUPACK development is funded by the National Science Foundation via the Molecular Programming Project and by the Beckman Institute at Caltech.
See also
External links
- NUPACK homepage
- Source download page
- Molecular Programming Project Homepage
- The Beckman Institute at Caltech
References
- ^ a b c d e Zadeh, J.N., C.D. Steenberg, J.S. Bois, B.R. Wolfe, A.R. Khan, M.B. Pierce, R.M. Dirks, and N.A. Pierce, NUPACK: analysis and design of nucleic acid systems. Journal of Computational Chemistry
- ^ a b Dirks, R.M., J.S. Bois, J.M. Schaeffer, E. Winfree, and N.A. Pierce, Thermodynamic analysis of interacting nucleic acid strands SIAM Review, 2007. 49(1): p. 65-88.
- ^ Serra, M.J. and D.H. Turner, Predicting thermodynamic properties of RNA. Methods in Enzymology, 1995. 259: p. 242-261.
- ^ Mathews, D.H., J. Sabina, M. Zuker, and D.H. Turner, Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. Journal of Molecular Biology, 1999. 288: p. 911-940.
- ^ SantaLucia, J., J., A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proceedings of the National Academy of Sciences of the United States of America, 1998. 95(4): p. 1460-1465.
- ^ SantaLucia, J. and D. Hicks, The thermodynamics of DNA structural motifs. Annual Review of Biophysics and Biomolecular Structure, 2004. 33: p. 415-440.
- ^ Koehler, R.T. and N. Peyret, Thermodynamic properties of DNA sequences: characteristic values for the human genome. Bioinformatics, 2005. 21(16): p. 3333-3339.
- ^ Dirks, R.M. and N.A. Pierce, A partition function algorithm for nucleic acid secondary structure including pseudoknots. Journal of Computational Chemistry, 2003. 24: p. 1664-1677.
- ^ Dirks, R.M. and N.A. Pierce, An algorithm for computing nucleic acid base-pairing probabilities including pseudoknots. Journal of Computational Chemistry, 2004. 25: p. 1295-1304.
- ^ a b Zadeh, J.N., B.R. Wolfe, and N.A. Pierce, Nucleic acid sequence design via efficient ensemble defect optimization. Journal of Computational Chemistry.
- ^ Dirks, R.M., M. Lin, E. Winfree, and N.A. Pierce, Paradigms for computational nucleic acid design. Nucleic Acids Research, 2004. 32(4): p. 1392-1403.
Categories:- Molecular modelling software

Wikimedia Foundation. 2010.